Human renal biopsy samples
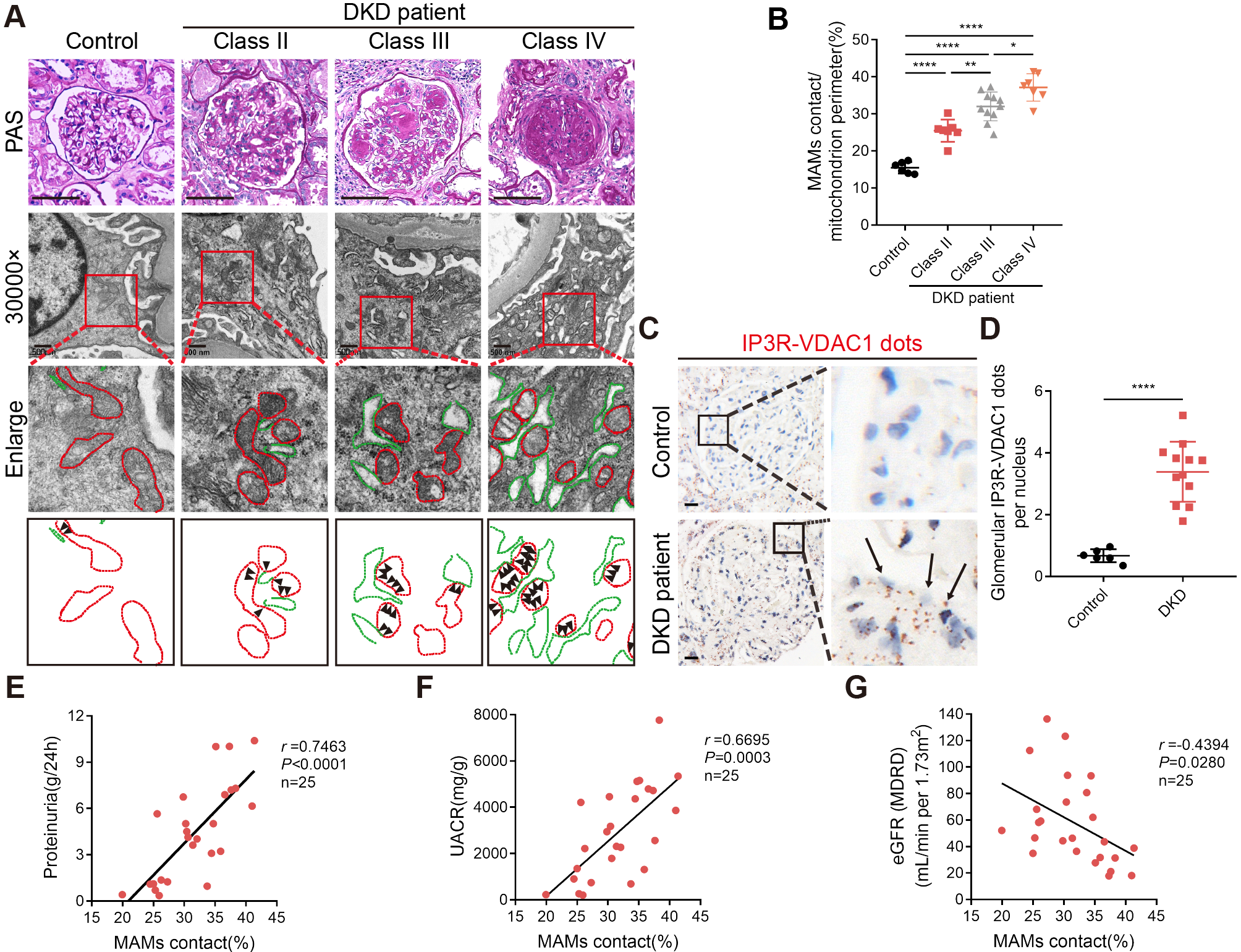
Renal biopsy samples from diabetic kidney disease (DKD) patients and normal kidney tissue (control group; adjacent healthy tissue from surgical resection of renal cell carcinoma samples without diabetes or other renal diseases) were obtained. DKD renal biopsy specimens were obtained from type 2 diabetes patients with a pathological diagnosis of DKD, accompanied by clinical syndrome characterized by persistent albuminuria (> 300 mg/24 h). All renal biopsy samples form patients diagnosed with DKD were classified in accordance with a new pathologic classification provided by the Renal Pathology Society. From mild to severe, DKD is divided into four hierarchical glomerular lesions as follows: Class I, glomerular basement membrane thickening: isolated glomerular basement membrane thickening and only mild, nonspecific changes by light microscopy that do not meet the criteria of classes II through IV. Class II, mild or severe mesangial expansion: glomeruli classified as mild or severe mesangial expansion but without nodular sclerosis (Kimmelstiel–Wilson lesions) or global glomerulosclerosis in more than 50% of glomeruli. Class III, nodular sclerosis (Kimmelstiel–Wilson lesions): at least one glomerulus with a nodular increase in the mesangial matrix (Kimmelstiel–Wilson) without changes described in class IV. Class IV, advanced diabetic glomerulosclerosis: global glomerulosclerosis in more than 50% of glomeruli with other clinical or pathologic evidence that sclerosis is attributable to DKD [34]. In this study, the renal specimens from patients with DKD we collected were from Class II- IV patients. The simplified (Chinese) MDRD equation was used to calculate the eGFR as follows: MDRD = 175× serum creatinine (mg/dL)−1.234 × years− 0.179 × (0.79, if female) [35]. This study was approved by the Ethics Committee of the Fifth Hospital of Sun Yat-Sen University (Zhuhai, China, 2022, No. K179-1), and all protocols were conducted in accordance with approved guidelines after informed patient consent was obtained. The basic clinical information of the human donors is shown in Supplementary Table S1.
Generation of podocyte-specific SGLT2 knockout mice and genotyping
C57BL/6J mice with an SGLT2 targeting vector were generated by Cyagen Biosciences Inc (Guangzhou, China). Homozygotes (SGLT2flox/flox) were generated by mating heterozygous (SGLT2flox/+) mice with each other. SGLT2flox/flox mice were further crossed with podocin promoter (NPHS2)-driven Cre recombinase transgenic (NPHS2-Cre+) mice in two successive breeding rounds to generate homozygous podocyte-specific SGLT2-deficient (SGLT2CKO) mice.
Mouse tail DNA was isolated using a Tissue Genomic DNA Extraction Kit (TIANGEN, Beijing, China) according to the manufacturer’s instructions. PCR was performed according to the PCR kit protocol (P131, Vazyme, Nanjing, China), and the primers used for PCR genotyping are shown in Supplementary Table S2. The sizes of the products were as follows. Flox 1: A 146-bp fragment was produced in the wild-type (WT) mice, and a 205-bp fragment was produced in SGLT2flox/flox or SGLT2CKO mice. Flox 2: A 172-bp fragment was produced in the WT mice, and a 238-bp fragment was produced in SGLT2flox/flox or SGLT2CKO mice. Cre: A 200-bp fragment was produced in SGLT2CKO mice, but no fragment was produced in WT or SGLT2flox/flox mice. The products were resolved on a 2% agarose gel using a TAE buffer system and a 50 bp DNA ladder (MD108, TIANGEN, Beijing, China) was used to determine the size of the products.
Animal protocol
The studies involving mice were approved by the Institutional Animal Care and Use Committee of the Fifth Affiliated Hospital of Sun Yat-Sen University (No.00314, Guangdong, China). Healthy four-week-old male wild-type C57BL/6J mice were purchased from Guangdong Medical Laboratory Animal Center (Foshan, Guangdong). Four-week-old podocyte-specific SGLT2 knockout male mice were generated as described above. All mice were maintained on a high-fat diet (HFD; 60 kcal% from fat, D12492, Research Diets) or a standard chow diet (12 kcal% from fat) and housed at 22–26 ℃ in a quiet room with a 12-hour light/dark cycle. After 4 weeks of HFD feeding, diabetic mice were induced by intraperitoneal injection of streptozotocin (STZ; S0130, Sigma-Aldrich, USA). Before receiving daily STZ injections, the mice were fasted for 12 h. STZ was intraperitoneally injected at a dose of 50 mg/kg body weight for 5 consecutive days. The control mice received an equal volume of 0.1 mol/L citrate buffer (pH = 4.5) at the same time. One week after the last injection, blood samples were taken from the tail vein of all mice to detect fasting blood glucose. Mice with fasting blood glucose levels ≥ 300 mg/dL were considered diabetic (marked as 0 week).
For the study of empagliflozin treatment in STZ/HFD-induced diabetic wild-type mice: Starting from the day when the diabetic model was confirmed to be successfully established through fasting blood glucose detection (marked as 0 week), and until the mice were euthanized (marked as 12 weeks), they were administered 10 mg/kg/d empagliflozin (HY-15409, MedChemExpress, USA) by daily oral gavage. The control mice also received an equal volume of 0.9% saline by daily oral gavage for 12 weeks.
All mice were anesthetized via isoflurane (RWD Life Science Co., China) inhalation and euthanized 12 weeks after the diabetic status was established. On the day before the mice were euthanized, they were housed in metabolic cages for 24 h and all urine was collected. Urine and serum were collected, and the kidneys and other organs were harvested for subsequent analysis.
Isolation of primary mouse podocytes
Mouse kidneys were removed and cut into pieces on ice, digested in culture medium containing collagenase A (COLLA-RO, Sigma-Aldrich, USA) and dispase (COLLDISP-RO, Sigma-Aldrich, USA) at 37 °C, and then passed through 100 μm and 70 μm sieves in succession. The filtered cells were coincubated with two podocyte-specific marker antibodies for 1 h (the antibodies used are shown in Supplementary Table S3), and then isolated by using streptavidin-conjugated M280 magnetic Dynabeads (Invitrogen, USA) [36, 37]. The magnetically attracted cells were considered to be primary podocytes and were processed for RNA or protein extraction.
Urine and serum analysis
Urine albumin was measured using an Albuwell M kit (1011, Exocell, Philadelphia, PA, USA), and the values were normalized to urine creatinine levels, which were quantified in the same samples with a urinary creatinine assay kit (1012, Exocell, Philadelphia, PA, USA) according to the manufacturer’s instructions. The urine albumin excretion rate was calculated as the urine albumin/creatinine ratio (UACR). Blood urea nitrogen was measured in serum using a Urea Nitrogen Colorimetric Detection Kit (EIABUN, Invitrogen, USA) according to the manufacturer’s instructions.
Histopathology analysis by light microscopy
Mice were perfused with 0.9% sodium chloride solution before kidney removal. Kidney tissues for light microscopy were immediately fixed in 4% paraformaldehyde (Biosharp, China) overnight, embedded in paraffin and further subjected to hematoxylin-eosin (H&E; DH0006, Leagene, Beijing, China), periodic acid-Schiff (PAS; DG0005, Leagene, Beijing, China) and Masson’s trichrome staining (DC0033, Leagene, Beijing, China) according to the manufacturer’s instructions. Light microscope analyses were based on 10 randomly selected glomeruli per mouse in each group using ImageJ software. The degree of glomerular mesangial expansion was assessed using PAS staining and the mesangial matrix index was quantified with ImageJ software as follows: mesangial matrix index = (mesangial area/total glomerular area)×100%. The mesangial area was defined as the PAS-positive and nucleus-free area, and the glomerular area was traced along the outline of the capillary loop and calculated using ImageJ. The assessment of glomerulosclerosis was based on the area of collagen deposition detected by Masson’s trichrome staining. The collagen deposition area was defined as the blue collagen-positive and nucleus-free area and calculated using ImageJ. The percentage of Masson’s trichrome staining area was quantified with ImageJ software as follows: Masson’s trichrome staining area = (collagen deposition area/total glomerular area)×100%.
Observation of glomerular basement membraney podocyte foot process MAMs and mitochondrial morphology by transmission electron microscopy
Renal tissue samples for electron microscopy were cut into 1mm3 sections and fixed with 2.5% glutaraldehyde (Alfa Aesar, USA) at 4 °C for 2 h. After washing, the tissue was post-fixed with 1% osmic acid (TED PELLA, USA) at 4 °C for 2 h. The tissue blocks were then dehydrated with ethanol and acetone and embedded in Epon812 Resin (TED PELLA, USA). Ultrathin 50–70 nm sections were cut using the cryo-ultramicrotome (Leica EM UC7, Leica, Germany), and then double-stained with 2% uranyl acetate (EMS, USA) and 3% lead citrate (TED PELLA, USA). Transmission electron microscope (JEM-1400 PLUS, Japan Electron Optics Laboratory Co., Ltd.) with an accelerating voltage of 100 kV was used to observe and photograph podocyte ultrastructure. Images were taken from randomly selected fields of view at 30,000× magnification. We obtained 3 different views for each sample.
ImageJ was used to quantify the average thickness of the glomerular basement membrane, and evaluate the average extent of foot process fusion and effacement.
MAMs and mitochondrial morphology were also assessed by transmission electron microscopy. Alterations in ER-mitochondrial contact points in podocytes were assessed using ImageJ software. MAMs were measured by the length of the mitochondrial membrane in contact with the ER (distance between the ER and mitochondria within 50 nm) and normalized to the mitochondrial perimeter, as previously described [38, 39]. Notably, some mitochondria in the images were not surrounded by the ER. In this case, the MAMs length would be recorded as 0. Then, we took the average of all the quantitative MAMs measured in all the random images as the final result for each sample. Besides, mitochondrial morphology was assessed using two mitochondrial fission-related parameters: mitochondrial aspect ratio and mitochondrial length. The mitochondrial aspect ratio was defined as the ratio of the major axis to the minor axis of the ellipse. The mitochondrial length was the length of the major axis. Then, we took the average of all the quantitative mitochondrial aspect ratio and mitochondrial length measured in all the random images as the final result for each sample.
SGLT2 immunofluorescence staining
The paraffin-embedded renal tissue sections and frozen renal tissue sections were sliced to approximately 5 μm thickness and mounted onto slides. The paraffin-embedded tissue slides were first deparaffinized and rehydrated. After antigen retrieval, the sections were blocked with donkey serum at 37 °C for 1 h and then incubated with primary antibodies at 4 °C overnight. The sections were washed 3 times in PBS and incubated with the secondary antibodies at 37 °C for 1 h on the next day. The primary antibodies and the secondary antibodies used for immunofluorescence are shown in Supplementary Table S3. DAPI staining was used to visualize the nuclei. Images were taken from randomly selected fields by confocal microscopy (LSM880, Carl Zeiss), and the mean fluorescence intensity was measured using ImageJ software.
Cell culture and treatment
Conditionally immortalized human podocytes, a gift from Prof. Saleem, were cultured as previously described [40]. Podocytes were incubated in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA) and 1% penicillin-streptomycin (Gibco, USA). Podocytes were cultured at 33 °C in 5% CO2 (permissive conditions) to induce cell proliferation.
To induce differentiation, the cells were maintained at 37 °C in 5% CO2 (non-permissive conditions) for at least 2 weeks. To prepare for the experiments, differentiated podocytes were exposed to normal glucose (NG, 5.5 mmol/L) or high glucose (HG, 30 mmol/L) medium for 72 h.
To further investigate the effects of mitochondrial calcium overload in MAMs-induced podocyte apoptosis, after incubation with NG or HG for 48 h, podocytes were treated with 100 nmol/L empagliflozin (HY-15409, MedChemExpress, USA) under NG or HG conditions for 24 h.
To further investigate the effects of empagliflozin, podocytes were pretreated with pretreated with BAPTA-AM (10 µmol/L, HY-100545, MedChemExpress, USA), or 2-APB (20 µmol/L, HY-W009724, MedChemExpress, USA), or Ru360 (10 µmol/L, 557440, Sigma-Aldrich, USA) for 1 h.
To investigate the roles of other selective SGLT2 inhibitors, after incubation with NG or HG for 48 h, podocytes were treated with 100 nmol/L canagliflozin (HY-10451, MedChemExpress, USA) or 100 nmol/L dapagliflozin (HY-10450, MedChemExpress, USA) under NG or HG conditions for 24 h.
In addition, to investigate the role of AMPK in SGLT2-mediated MAMs formation, after incubation with NG or HG for 48 h, podocytes were treated with 1 mmol/L of the AMPK activator AICAR (HY-13417, MedChemExpress, USA), or 5 µmol/L of the AMPK inhibitor Compound C (HY-13418, MedChemExpress, USA), or 100 nmol/L of empagliflozin together with 5 µmol/L of Compound C under NG or HG conditions for 24 h.
Small interfering RNA (siRNA) and plasmid transfection
SGLT2 (also known as SLC5A2) siRNA was purchased from HanYi Bio (Guangzhou, China). The siRNAs used in this study were as follows: SGLT2 sense, AGAAGGCCCUGAUUGACAATT; SGLT2 anti-sense, UUGUCAAUCAGGGCCUUCUTT. The siRNAs were dissolved in ddH2O to prepare a 20 µmol/L stock solution. Differentiated podocytes grown in 6-well plates were transfected with siRNA in transfection medium containing Lipofectamine 3000 transfection reagent (Invitrogen, USA). For each transfection, 100 µL of transfection medium containing 5 µL of siRNA stock solution was gently mixed with 100 µL of transfection medium containing 5 µL of transfection reagent. After incubation for 15 min, the complexes were added to podocytes in 1.8 mL of transfection medium. Podocytes were incubated with this mixture for 6–8 h at 37 °C, after which the transfection medium was replaced with normal medium.
SGLT2 (also known as SLC5A2)-3×Flag plasmid was purchased from HanYi Bio (Guangzhou, China) and constructed using the pcDNA3.1 vector. The pcDNA3.1-3×Flag negative control plasmid was used as a control. OMM-ER linker plasmid (also known as OMM-RFP-ER, a synthetic linker encoding a monomeric red fluorescent protein (RFP) between the N-terminal outer mitochondrial membrane (OMM) mAKAP1 targeting sequence and the C-terminal ER yUBC6 targeting sequence) and DsER plasmid (control plasmid) were gifts from Prof. Liu [41, 42]. Based on the size of the fluorescent protein (4.2 × 2.4 nm), the maximal length of this construct was < 5 nm. Therefore, this construct can decrease the distance between these organelles and increase the number of ER-mitochondrial contact sites [41, 42]. Podocytes grown in 6-well plates were transfected with plasmid in transfection medium containing Lipofectamine 3000 (Invitrogen, USA). For each transfection, 100 µL of transfection medium containing 2 µg of plasmid and 4 µL of p3000 was gently mixed with 100 µL of transfection medium containing 4 µL of transfection reagent. After incubation for 15 min, the complexes were added to podocytes in 1.8 mL of transfection medium. Podocytes were incubated with this mixture for 6–8 h at 37 °C, after which the transfection medium was replaced with normal medium.
In situ proximity ligation assay (PLA)
Labeling the classical MAMs structure of the Inositol-1,4,5-triphosphate receptor (IP3R) - Glucose regulated protein 75 (GRP75) - Voltage-dependent anion channel 1 (VDAC1) complex by in situ PLA is a classical method for identifying MAMs and has been widely used in the detection and quantification of MAMs in the last decade [39, 43,44,45]. In this study, the status of MAMs was evaluated using IP3R - VDAC1 PLA assays. The primary antibodies used were anti-IP3R and anti-VDAC1 (the details of the antibodies are shown in Supplementary Table S3). Immunofluorescent IP3R-VDAC1 PLA assays were performed using the Duolink® In Situ Detection Kit according to the manufacturer’s instructions (DUO92002, DUO92004, DUO92008 or DUO92014, Sigma-Aldrich, USA). Besides, immunohistochemical IP3R-VDAC1 PLA assays were done using the Duolink® In Situ Detection kit according to the manufacturer’s instructions (DUO92002, DUO92004, DUO92012, Sigma‐Aldrich, USA). Images were taken from randomly selected fields of view using confocal microscopy (LSM880, Carl Zeiss, Germany) or Olympus BX53 microscope (Olympus, Japan). PLA signals were quantified using the “Particle Analysis” function of ImageJ software.
Flow cytometric analysis of apoptosis
Podocytes (2 × 105 cells/well) were harvested using 0.25% Trypsin without EDTA for 4 min. After double-staining with Annexin V-FITC and propidium iodide (PI) according to the instructions of the Annexin V-FITC/PI Apoptosis Detection Kit (A211, Vazyme, Nanjing, China), the cells were subjected to flow cytometry (CytoFlex LX, Beckman, USA). A total of 105 cells were analyzed for each sample, and each condition was tested in three independent samples. FlowJo v10 software was used to calculate the percentage of apoptotic cells. The apoptosis rate = Q3 (Annexin V-FITC+/PI–, early apoptotic cells) + Q2 (Annexin V-FITC+/PI+, late apoptotic cells).
TUNEL apoptosis assays
A TUNEL BrightGreen Apoptosis Detection Kit (A112, Vazyme, Nanjing, China) was used based on the manufacturer’s protocols. Briefly, frozen renal sections were dried at room temperature for 20 min and then fixed in 4% paraformaldehyde for 30 min. The sections were incubated with 0.2% Triton X-100 for 15 min and equilibrated with equilibration buffer for 30 min, followed by incubation with 50 µL of the TdT reaction mixture at 37 °C for 60 min in the dark. Subsequently, the sections were counterstained with DAPI. Images were taken from randomly selected fields of view using confocal microscopy (LSM880, Carl Zeiss, Germany). The number of apoptotic cells in the glomeruli for each sample was counted for quantification.
Measurement of calcium fluorescence
Specific calcium fluorescent probes Mag-Fura-2 AM (2 µM, Invitrogen, USA) and Rhod-2 AM (5µM, Invitrogen, USA) were used to measure the ER and mitochondrial Ca2+ concentrations, respectively. Podocytes were co-incubated with Mag-Fura-2 AM and Rhod-2 AM in calcium-free Hanks’ Balanced Salt Solution (Biosharp, China) at 37 °C for 30 min, and then washed to remove the extraneous dye. Hoechst 33,342 (Beyotime, China) was used to stain nuclei. Cells were immediately measured ER and mitochondrial Ca2+ concentrations using confocal microscopy (LSM880, Carl Zeiss, Germany) at 458 nm and 543 nm excitation wavelengths, respectively. Image mean fluorescent intensity was quantified using Image J software.
Isolation of MAMs from podocytes
The MAMs, mitochondrial, and ER fractions were isolated from cultured podocytes by Percoll gradient fractionation. Briefly, podocytes from twenty 100 mm cell culture dishes were digested with trypsin and centrifuged at 600 ×g for 5 min twice to collect the cells. Then, the podocytes were lysed in buffer 1 (225 mM mannitol, 75 mM sucrose, 0.1 mM EGTA and 30 mM Tris-HCl, pH = 7.4) supplemented with protease inhibitors. The homogenate was centrifuged twice at 600 ×g for 5 min to collect the supernatant. The supernatant was then recentrifuged for 10 min at 7,000 ×g to separate the resulting pellet (crude mitochondrial fraction) and supernatant (ER fraction). Using a 41.1 SW rotor in the Beckman ultracentrifuge, the supernatant was centrifuged at 100,000 ×g for 1 h to obtain the resulting pellet (ER fraction). The crude mitochondrial fraction was resuspended in buffer 2 (225 mM mannitol, 75 mM sucrose and 30 mM Tris-HCl, pH = 7.4) and centrifuged twice at 7,000 ×g for 10 min to collect the resulting pellet. Then, the pellet was resuspended using a 30% Percoll gradient in mitochondrial resuspending buffer (MRB; 250 mM mannitol, 5 mM HEPES pH = 7.4 and 0.5 mM EGTA) and centrifuged at 95,000 ×g for 30 min. The pure mitochondrial fraction was obtained at the bottom of the tube, and the crude MAMs fraction was localized to the intermediate layer. Then, the pure mitochondrial fraction was resuspended in MRB buffer and centrifuged twice at 6,300 ×g for 10 min to collect the resulting pellet (mitochondrial fraction). The crude MAMs fraction was also resuspended in MRB buffer and centrifuged at 6,300 ×g for 10 min to collect the supernatant. Then, the supernatant was centrifuged at 100,000 ×g for 1 h to collect the resulting pellet (MAMs fraction) [38, 46]. Equal amounts of protein (25 µg) from each fraction were used for further western blot analysis.
Adenosine diphosphate (ADP)/ adenosine triphosphate (ATP) ratio assay
Podocytes were grown and differentiated on a white 96-well plate at a density of 3000–4000 cells per well. The ADP/ATP ratio was measured via an ADP/ATP Ratio Assay Kit-Luminescence (A552, Dojindo, Japan) according to the manufacturer’s protocols.
Quantitative real-time PCR
Total RNA from cultured human podocytes was obtained using a Total RNA Kit I (R6834, Omega, Guangzhou, China). Total RNA was isolated from mouse tissue using TRIzol reagent (TaKaRa Biotechnology, Dalian, China). RNA purity and concentration were measured using a NanoDrop 2000 (Thermo Fisher Scientific). cDNA was prepared using the PrimeScript RT Reagent Kit (RR047A, TaKaRa Biotechnology, Dalian, China) according to the manufacturer’s instructions. Real-time PCR was performed in a CFX96 Touch Real-time PCR Detection System (Bio-Rad, USA) with 12.5 µL of TB Green Premix Ex Taq II (Tli RNaseH Plus) (RR820A, TaKaRa Biotechnology, Dalian, China), 0.4 µM primers, 2 µL of cDNA and 8.5 µL of ddH2O. The cycling conditions were as follows: 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Relative gene expression was normalized to that of the β-actin internal control and calculated using the 2−ΔΔCq method in Microsoft Office Excel. The primers used for quantitative real-time PCR are shown in Supplementary Table S2.
Western blot analysis
Podocytes were lysed in RIPA buffer (Beyotime Technology, Shanghai, China) containing protease and phosphatase inhibitor cocktail (Beyotime Technology, Shanghai, China) at 4 °C. The suspension was centrifuged at 12,000×g and the supernatant containing the proteins was collected. A total of 30 µg of protein lysate was diluted in loading buffer, separated by 10% SDS‒PAGE (EpiZyme, Shanghai, China), and transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories, Hercules, CA, USA) at 100 V for 2 h. The membranes were rinsed in Tris-buffered saline with Tween 20 (TBST, Beyotime Technology, Shanghai, China) for 5 min three times. Then, the membranes were immersed in blocking buffer (5% milk powder in TBST) for 1 h and then rinsed in TBST for 5 min three times. The membranes were incubated with the primary antibodies overnight at 4 °C. After rinsing in TBST for 5 min three times, membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature and developed using enhanced chemiluminescence (ECL) reagent (WBKLS0500, Merck Millipore, USA). The primary antibodies and the secondary antibodies used are shown in Supplementary Table S3. Densitometric analyses were conducted using ImageJ software.
Statistics
The data were expressed as the mean ± standard deviation, and all experiments were performed at least three independent times, unless stated otherwise. The data were evaluated with GraphPad Prism (version 9.0, GraphPad Software, La Jolla, CA, USA). Comparisons between two groups were performed by two-tailed Student’s t tests. Comparisons among multiple groups were tested using one-way or two-way ANOVA with a multiple comparisons post hoc test. A normal distribution was assessed using a Kolmogorov‒Smirnov test and normal data distribution was assumed when P > 0.05. Non-normal distributed data were analyzed by Mann‒Whitney test. Differences were considered significant at P < 0.05.






留言 (0)