Antibodies, reagents, buffers, and solutions
The antibodies, special reagents, buffers and solutions used are listed in Tables 1 and 2.
Table 1 Antibodies and reagentsTable 2 Buffer and solutionsCell culture and cell stimulation
A549 and A431 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), and HEK293 cells were cultured in minimum essential medium (MEM) supplemented with 10% FCS. HEK-µOR TRPC4α [15] cells were cultured in DMEM supplemented with 10% FCS, 100 µg/mL hygromicin B, and 500 µg/mL G418. HEK-TRPM3α2 [16] and HEK-CCE2 [17] (TRPC5) cells were cultured in MEM supplemented with 10% FCS and G418 (500 and 400 µg/mL, respectively). All the cells were incubated at 37 °C in a 5% CO2 atmosphere. For stimulation experiments, cells were seeded in 6-well plates and grown until they reached confluence. The cells were starved with 0.5% FCS media overnight, followed by an incubation in FCS-free media for 1 h and stimulation as indicated in the figure legends. For ADAM10 inhibition, a preincubation step with 10 µM GI254023X for 30 min was included prior to stimulation. The cells were subsequently lysed with ADAM10 lysis buffer, followed by centrifugation for 15 min at 16,000 x g and storage of the supernatant at -80 °C.
Western blot analysis and E-cadherin cleavage calculation
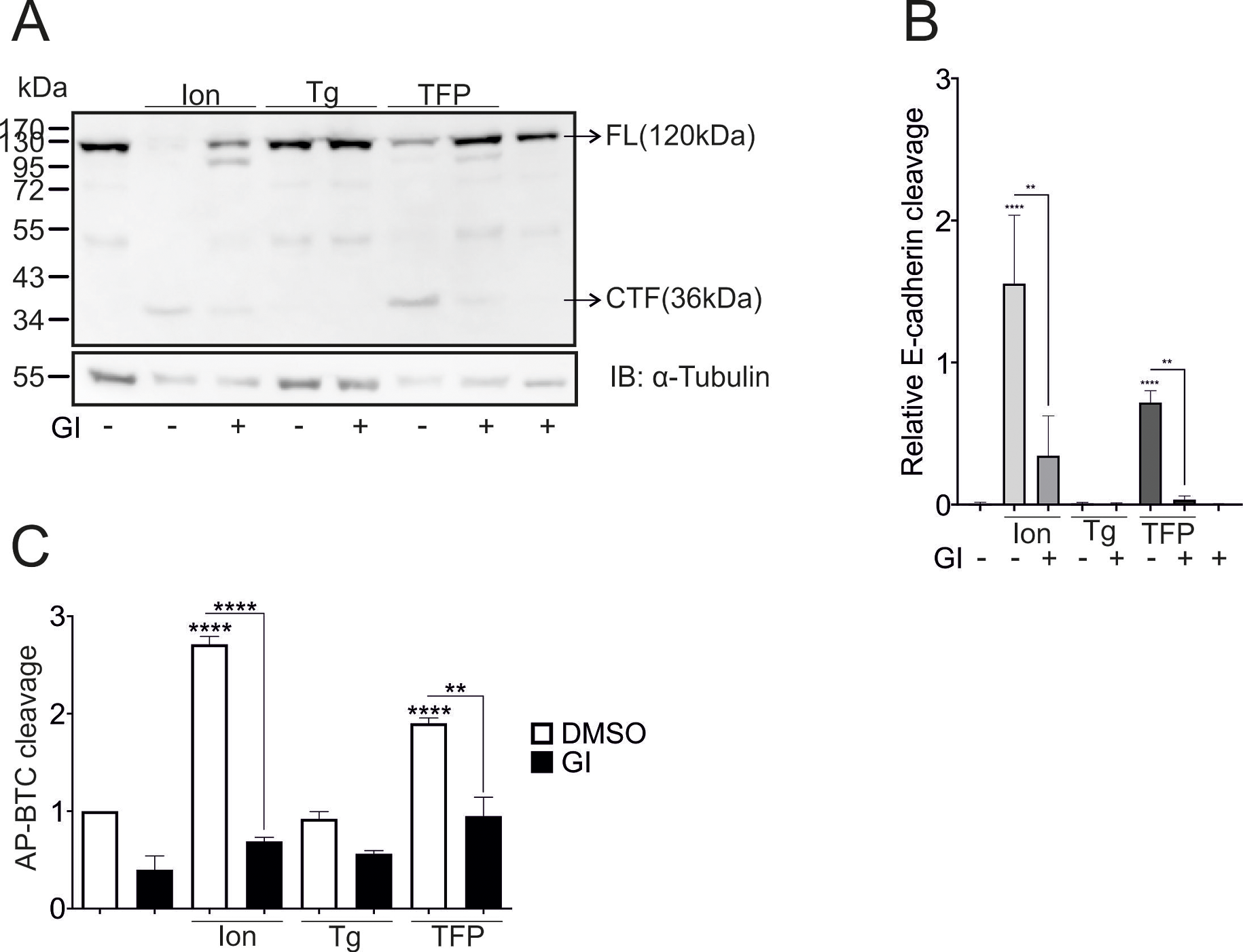
The protein concentration of the cell lysates was quantified using a bicinchoninic acid (BCA) assay kit (Thermo Fisher, Karlsruhe, Germany) according to the manufacturer’s protocol. The samples were heated in SDS buffer at 60 °C for 30 min before being subjected to SDS‒PAGE (10% Tris‒glycine gels), in which equal protein amounts were loaded per lane and blotting onto nitrocellulose membranes (Amersham Protran Premium 0.45 NC, GE Healthcare Life Sciences, Freiburg, Germany). The membranes were blocked with 5% nonfat milk in Tris-buffered saline containing 0.05% Tween for 1 h, incubated with the primary antibody overnight (Table 1) and then incubated with the corresponding secondary antibody for 1 h at RT. The chemiluminescence substrate (PerkinElmer, Waltham, MA, USA) was added to the membranes, and the chemiluminescent signals were recorded using a LAS3000 device (Fujifilm, Tokyo, Japan). AIDA Image Analysis software 4.27.039 (Elysia-raytest, Angleur, Belgium) was used for image acquisition and densitometric analysis after background subtraction utilizing equal square sizes for the bands of interest. For the determination of E-cadherin cleavage activity, membranes were developed using an anti-mouse C-terminal E-cadherin antibody, and the densitometry analysis was performed by calculating the ratio of the 36 kDa C-terminal fragment (CTF) to the 120 kDa full-length (FL) protein with the following formula: \(\:\frac\). The results were normalized to the amount of protein loaded onto the gels.
Coimmunoprecipitation
HEK-µOR TRPC4, HEK-TRPM3α2 and HEK-CCE2 cells were lysed in IP lysis buffer and equilibrated at 4 °C for 30 min. Subsequently, the lysates were centrifuged for 45 min at 100,000 x g, and the supernatants were incubated with antibody-coupled beads overnight at 4 °C. For the preparation of the antibody-coupled beads, 60 µL of Protein G Dynabeads (beads) per sample was washed twice with IP lysis buffer (magnetic stand). The appropiate amount of beads was incubated with 10 µg of TRPC4, TRPC5, TRPM3 or ADAM10 C-terminal antibody in of 500 µL IP lysis bufferfor 1 h, and washed five times with IP lysis buffer to remove the unbound antibody. After the overnight incubation of the beads and lysates, the beads were collected (magnetic stand) and denatured in SDS buffer for 20 min at 60 °C. The samples were then subjected to Western blot analysis developing with antibodies against TRPC4, TRPC5, TRPM3 or the ADAM10 C-terminus as described above.
Antibody surface staining and flow cytometry
A549 and A431 cells were dissociated with accutase (Merck Millipore, Darmstadt, Germany), resolved in FACS buffer and centrifuged for 5 min at 300 x g. The cells were kept on ice throughout the following steps. The supernatant was removed, and the cells were incubated with an ADAM10 MAB004 antibody (1 µg/mL), isotype control (1 µg/mL) or FACS buffer for 1 h. The specificity of the ADAM10 antibody was reported in a previous publication with ADAM10 deficient cells [18]. The cells were washed twice and incubated with the secondary anti-mouse antibody A-21235; (2 µg/mL) for 45 min. Afterwards, the cells were washed twice, fixed in 1% paraformaldehyde (PFA) for 5 min and finally diluted with 0.1% PFA for fluorescence signal detection using the Sony SH800 (Sony, Berlin, Germany) and FACS Aria Fusion, BD, Heidelberg, Germany) systems. An equal amount of cells was recorded per sample. The data were analyzed with FlowJo 10.6.2 software (Tree Star, Inc., Ashland, OR, USA). Cells with decreased viability and cell debris were excluded by forward and side scatter gating, and the mean fluorescence of the samples minus the mean fluorescence of the isotype control was used for comparison.
Alkaline phosphatase (AP) assay
A549 cells were transfected with a plasmid encoding N-terminally coupled alkaline phosphatase (AP)-betacellulin fusion protein using Lipofectamine™ 3000 (Thermo Fisher, Karlsruhe, Germany) and seeded in 12-well plates at a density of 3*105 cells per well [19]. After the respective treatments and sampling, 100 µl of the cell culture supernatant and the corresponding cell lysate (in AP-assay lysis buffer) was transferred to a 96-well plate, and 100 µl of a 2 mg/ml 4-nitrophenyl phosphate solution (AP substrate, resolved in AP-assay activity buffer) was added to each well. Importantly, the plate must be kept cool/on ice before the start of the measurement to avoid unintended substrate turnover. Protease activity was quantified as equivalent substrate turnover by AP by measuring of the absorption at 405 nm every 1.5 min for 2.5 h at 37 °C using Genios fluorescence reader (Tecan, Grödig, Austria). Finally, the slopes of the linear region of the curve were determined, and the ratio of the activity in the supernatant to the activity in the lysate plus the supernatant (total activity) was calculated to obtain the final activity in the sample.
Exosome preparation
A total of 2 × 107 A549 cells were seeded in 100 mm dishes, and after adherence, the cells were cultured overnight in DMEM containing 0.5% FCS. The cells were further starved with TBS with or without Ca2+ followed by ionomycin or trifluoperazine stimulation. The medium was collected and subjected to differential centrifugation (10 min at 300 x g, 20 min at 1,000 x g, 30 min at 10,000 x g). After each centrifugation step, the pellets were lysed with SDS buffer, and the supernatants were subjected to the next centrifugation step. The supernatants were filtered through a 0.22 μm membrane filter, and the extracellular vesicles were collected by centrifugation at 100,000 x g for 1 h at 4 °C using a Beckman rotor Type Ti50.2. The pellet was washed with ice-cold PBS and then centrifuged at 100,000 x g for 1 h. Vesicles were directly lysed with SDS-sample buffer for Western blot analysis or further fractionated by gradient centrifugation for pure exosome preparation as described previously [12]. Briefly, the vesicle pellets were resuspended in PBS and loaded onto a continuous sucrose gradient (2, 1.3, 1.16, 0.8, 0.5 and 0.25 M sucrose). After centrifugation at 100,000 x g for 16 h at 4 °C (Beckman Ti50.2 rotor), six 1 ml fractions were collected, and the density of each fraction was measured and subsequently centrifuged at 150,000 x g for 4 h at 4 °C using a Beckman TLA-55 rotor. The pellets were collected and subjected to Western blot analysis.
Measurement of cytoplasmic Ca2+ levels with Fura-2
A549 cells (2.5*105) were seeded on poly-L-lysine (Sigma, Steinheim, Germany) coated glass coverslips and grown until 70% confluence. The cells were loaded with 4 µM Fura-2 AM (Invitrogen by Thermo Fisher Scientific, Eugene, Oregon, USA) for 30 min at 37 °C and then washed with Ringer’s solution with or without Ca2+. The coverslips were placed into a circular open-bottom chamber, and washed again before the chamber was fitted onto the stage of an inverted microscope (Axivert S100, Carl Zeiss, Jena, Germany) with a monochromator (Polychrome V, TILL Photonics, Germany). The measurements were performed at room temperature. Every 2 s, the fluorescence from Fura-2 was captured by alternating excitation (0.5 Hz) at 340 and 380 nm for 20 ms, while the emission was recorded with a cooled charge-coupled device (CCD) camera (Imago, TILL Photonics). The cells were marked as regions of interest (ROIs) whereas two to three empty areas were selected as backgrounds to be subtracted. The changes in the ratios of single ROIs were recorded, and the F340/380 ratios versus time were plotted automatically. Individual cells with significant deviations were considered outliers based on the observations of the XY ratio. Monochromator operator, image acquisition, and analysis were controlled by TillvisION software (TILL Photonics, Planegg/Martinsried, Germany). The final results are shown as the F340/F380 ratio, area under the curve or amplitude.
Fura-2 AM calibration and Ca2+ concentration calculation
The cells were seeded and treated as described above. After the washing steps, Ringer’s solution was removed and replaced with 160 µL of 0Ca/EGTA solution, and the imaging experiment for the cytoplasmic Ca2+ measurement was started. When the signal plateau was reached, the Rmin (F340/380 value) and Sf2 (excitation intensity of the 380 nm channel in Ca2+ free conditions) values were extracted. A total of 240 µL of 10Ca/EGTA solution was added resulting in a final Ca2+ concentration of 110 nM (calculated using the webmaxc standard). In the plateau phase, the R110nM/Ca value was extracted. Finally, 400 µL of 20Ca saline solution was added, and Rmax and Sb2 (excitation intensity of the 380 nm channel in excess Ca2+ conditions) values were extracted. Five independent measurements with a total of 170 cells were performed The following equation was used to calculate the experimental dissociation constant:
$$\:_=110nM/\left(\frac_-_}_-_}*\left(\frac\right)\right)$$
The Ca2+ concentrations from other Ca2+ imaging experiments were calculated using the following equation
$$\:\left[^\right]i=_*\frac_-_\text\text}}_-_}*\left(\frac\right)$$
as described earlier with the dissociation constant value [20].
sADAM10/exosome related ADAM10 FRET-based activity assay
Soluble or exosome related ADAM10 activity in the supernatant was assessed with the FRET substrate PEPDab063A (Dabcyl-HGDQMAQKSK(5FAM)-NH2) (Biozyme Inc., North Carolina, USA). This substrate contains a consensus cleavage sequence with high specificity for ADAM10 [21]. Upon cleavage, the quencher is released from the fluorophore resulting in an increase in fluorescence. A549 and A431 cells were seeded in 12 well plates, starved overnight in DMEM supplemented with 0.5% FCS, and finally equilibrated in phenol red-free and serum-free DMEM for one hour. After the indicated treatments and sampling, the supernatant was centrifuged, diluted 1:2 in phenol red-free DMEM (50 µL end volume), and added to a 96 well reading plate in triplicate together with a positive control (trypsin) and a negative control containing a 1:1 mixture of ADAM10 FRET-based activity assay buffer and phenol red-free DMEM. The GI inhibitor was directly added to the plates. Importantly, the plate must be kept cool/on ice to avoid unintended substrate cleavage. The FRET-substrate was added to a final concentration of 10 µM, and fluorescence was measured in a TECAN plate reader (Zürich, Switzerland) every 2 min for 4 h with a gain optimization function (excitation wavelength: 485 nm; emission wavelength: 530 nm). For data acquisition, the average of the technical replicates was obtained, and the slope of the linear region of the curve was calculated for each sample. The results were normalized to the experimental control.
Statistical analysis
The quantitative data are shown as the means + SD from three independent experiments, unless indicated otherwise. The statistical analysis was performed using GraphPad PRISM 9.0 (GraphPad Software, La Jolla, CA). A p-value < 0.05 was considered significant. The statistical analysis of the nonnormalized E-cadherin cleavage data was performed using ANOVA followed by a Tukey’s post- hoc test for multiple comparisons between groups. Significant differences in AP-BTC cleavage (normalized data) were analyzed with a one-sample t-test followed by a false discovery rate (FDR) analysis. For details on the other experiments see the figure legends.






留言 (0)