
記住我





Five New Zealand white male rabbits (3.5–4.0 kg; Nippon Bio-Supp. Center, Tokyo, Japan) were utilized to obtain blood/bone marrow concentrates and primary culture cells in the present study. All the animals were treated without excessive or disruptive noise and were fed a standard diet and water ad libitum at the Central Animal Care Facility at XXX University. This study was approved by the Animal Experiment Ethics Committee of XXX University (Approved No. 20-08-01).
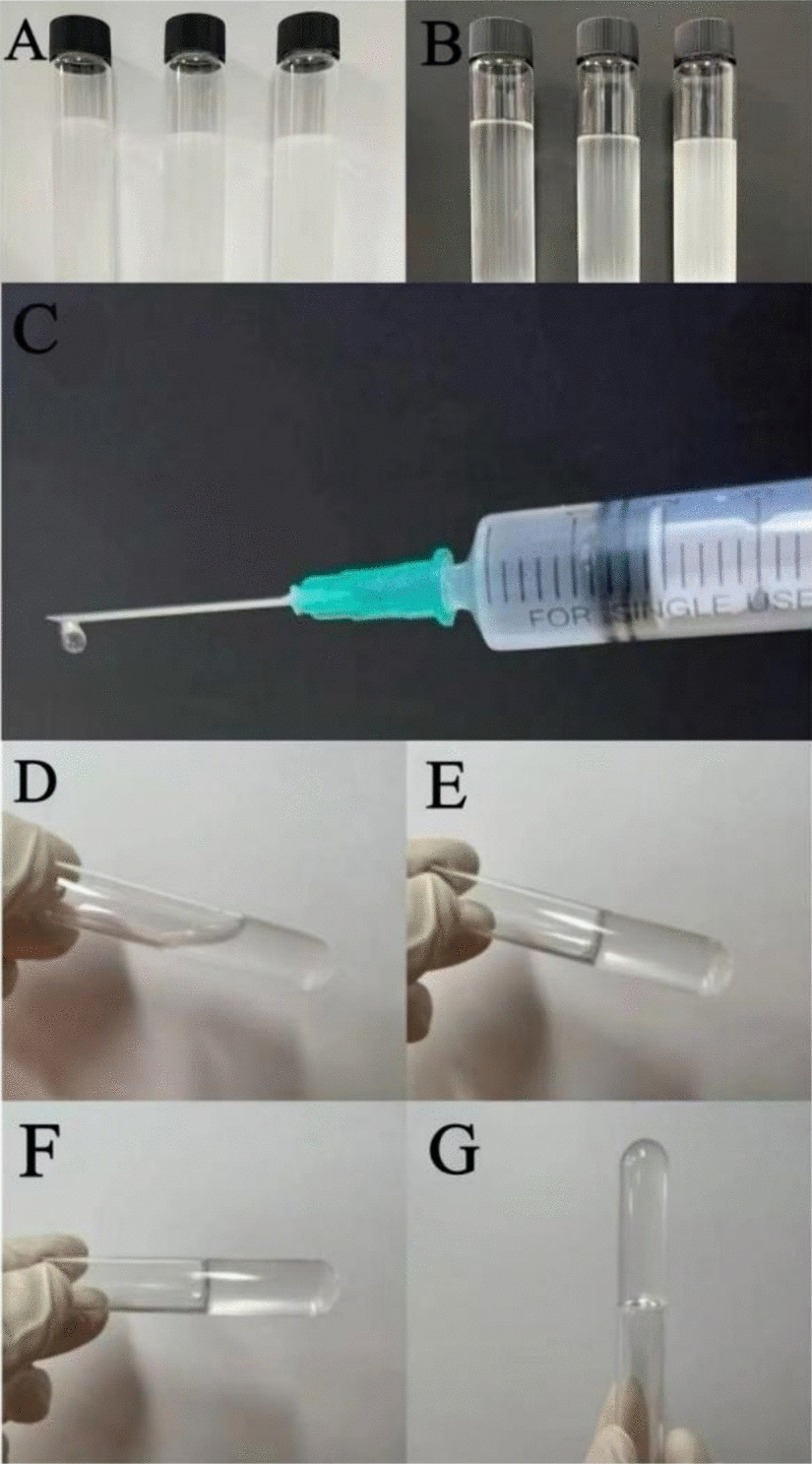
General anesthesia was applied by intramuscular injection of a mixture of butorphanol tartrate (Meiji Seika Pharma Co., Ltd., Tokyo, Japan), medetomidine hydrochloride (Nippon Zenyaku Kogyo Co., Ltd., Fukushima Japan), and midazolam (Astellas Pharma, Inc., Tokyo, Japan). Arterial blood was collected from the auricular arteries, and bone marrow aspirate was collected from the iliac bones. Each 2.5-mL blood or bone marrow aspirate was centrifuged in a plastic tube without anticoagulant at 700×g for 5 min at room temperature in a horizontal centrifuge machine (LC-200, TOMY, Tokyo, Japan). After centrifugation, the upper layer of components with the buffy-coat layer site and an approximately 5-mm red blood cell layer were collected as iPRF or iBMAC (Fig. 1A, C). The 5-mm red blood cell layer includes more cellular components releasing growth factors [13, 14] and this fraction should be included for clinical use [15].
Fig. 1
A–D Visual representation of layer separation of injectable platelet-rich fibrin (iPRF) or injectable bone marrow aspirate concentrate (iBMAC). (A, C) The separated layers after centrifugation of blood/bone marrow aspirate. (B, D) The iPRF or iBMAC generated from cell culture plastics. E–H Histological observation of frozen sections of each concentrate material. (E, F) The general parts of iPRF and iBMAC. More hematoxylin-stained nucleated cells were observed in the iBMAC specimens than in the iPRF specimens. (G) Aggregated clusters of cells containing leukocytes were occasionally observed in the iPRF specimens. (H) Many hematoxylin-stained cells, including leukocytes, adipocytes, and other bone marrow stromal cells, were located within and around the cluster of platelets and adipose cells in iBMAC
2.2 Histological sample preparationThe collected iPRF and iBMAC cells were transferred to cell culture plastic dishes and clotted within 10 min (Fig. 1B, D). The clotted specimens were fixed in 4% formaldehyde (Nacalai Tesque, Kyoto, Japan) in PBS and embedded in supercryoembedding medium (SCEM, Section-Lab, Hiroshima, Japan). The frozen specimens were sectioned into 8-μm-thick slices in the chamber of a cryomicrotome (Leica CM3050S, Leica, Tokyo, Japan) with adhesive film (Cryofilm Type 2C (16UF), Section-Lab) as previously reported [11, 16]. Images of the sections stained with hematoxylin were captured with an AXIO Imager M2 microscope (Carl Zeiss, Jena, Germany).
2.3 Scanning electron microscopy (SEM)The prepared iPRF and iBMAC were fixed overnight at 4 °C using a 2.5% glutaraldehyde solution (Sigma Aldrich, St. Louis, MO, USA), dehydrated in an elevated ethanol series and dried with hexamethyldisilazane (Sigma Aldrich). The dried specimens were attached to aluminum stubs (Nisshin-EM, Tokyo, Japan) and sputter-coated with platinum with a sputtering device (E-1030 Ion Sputter, HITACHI, Tokyo, Japan). SEM images were acquired using a scanning electron microscope (SEM; JSM-IT200, JEOL, Tokyo, Japan).
2.4 Cell cultureEither iPRF or iBMAC was incubated in 10 mL of DMEM (Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 1% antibiotics (Gibco) at 37 °C. After 24 h, the incubated medium (conditioned medium) was collected and stored at − 20 °C until use. When added to the culture medium, both iPRF and iBMAC gradually coagulated within 10–15 min in the media. Therefore, the conditioned media did not contain the iPRF or iBMAC in their original forms; rather, they contained the growth factors released from these materials.
Rabbit primary gingival fibroblasts (RGFs) and rabbit primary osteoblasts (ROBs) were isolated and cultured from gingival tissue and bone chips from the mandible of rabbits, respectively, as previously described [11]. Since rabbits were selected as the source of the blood and bone marrow in the present study, rabbit gingival fibroblasts (RGFs) and osteoblasts (ROBs) were isolated to test the effects of iPRF and iBMAC from the same animals. Both cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% antibiotics in cell culture flasks at 37 °C in a humidified atmosphere. The cells were detached from the tissue culture plastic with 0.25% EDTA-trypsin (Gibco) before reaching confluency. The cells from passages 4–6 were used for experimental cell seeding.
The cells were seeded in medium supplemented with 20% conditioned medium (or 20% control medium without FBS) and 80% basic cell culture medium (DMEM supplemented with 10% FBS and 1% antibiotics) at a density of 10,000 cells for a live/dead cell assay, 50,000 cells for collagen 1 (COL1) staining per well on 8-well chamber slides (IWAKI, Sizuoka, Japan), and 50,000 cells for real-time PCR and alizarin red staining assays per well in 24-well plates.
2.5 Live/dead cell assayTwenty-four hours after cell seeding, the cells were evaluated using a live/dead staining assay according to the manufacturer’s protocol (Live-Dead Cell Staining Kit, Enzo Life Sciences, Farmingdale, NY, USA). Fluorescence images were quantified with an inverted fluorescence microscope (Olympus FSX-100). The cell quantities are expressed as the percentage of live-to-dead cells.
2.6 Migration assayA migration assay was performed using polyethylene terephthalate cell culture inserts with an 8-μm pore size (Millicell cell culture insert, Merck Millipore, MA, USA) in a 24-well plate. The lower compartment of each well was filled with medium supplemented with 20% conditioned media and 80% basic cell culture media. After starvation with DMEM containing 0.5% FBS for 12 h, 10,000 resuspended cells were seeded in the upper compartment. After 24 h of incubation, the cells were fixed with 4% formaldehyde for 2 min. Then, the cells were permeabilized with acetone for 15 min and stained with Giemsa solution for 20 min. The upper side of the filter membrane was rinsed and gently wiped with a cotton swab to remove cell debris. Images of the migrated cells were captured, and the number of cells under the filter was counted under a microscope (AXIO Zoom. V16, Carl Zeiss).
2.7 Real-time PCR analysisTotal mRNA was isolated from RGFs at 3 and 7 days and from ROBs at 3 and 14 days poststimulation with conditioned medium to determine the relative mRNA levels of TGF-β, VEGF, and COL1 in the RGFs and runt-related transcription factor 2 (Runx2), COL1, and osteocalcin (OCN) in the ROBs. The specific time points of days 3 and 7 for RGFs, and days 3 and 14 for ROBs, were chosen based on the results from a similar previous study [11]. The sequences of primers used for the genes were generated according to the information presented in Table 1. mRNA isolation was performed using a ReliaPrep RNA Cell Miniprep system (Promega, Madison, WI, USA), and real-time PCR was performed using a GoScript reverse transcription system (Promega) and quantified on a StepOne Plus PCR system (Applied Biosystems, Waltham, MA, USA). The ∆∆Ct method was used to calculate gene expression levels normalized to the expression of β-actin.
Table 1 List of primer sequences for real-time PCR2.8 Collagen immunofluorescence stainingFourteen days after seeding with conditioned medium, RGFs were investigated for COL1 expression by using immunofluorescence staining. The time point was chosen based on the previous work [11]. The RGFs were fixed with 4% formaldehyde for 10 min, permeabilized with PBS containing 0.2% Triton X-100, and blocked with PBS containing 1% bovine serum albumin (BSA; Sigma Aldrich) for 1 h. Subsequently, the cells were incubated overnight at 4 °C with an anti-mouse monoclonal COL1 antibody (MA1-26771; Invitrogen, Waltham, MA, USA) diluted 1:200 in PBS containing 1% BSA. After the cells were washed with PBS, they were incubated for 1 h at room temperature with secondary antibodies (goat anti-mouse Alexa Fluor Plus 488, Invitrogen) diluted 1:200 in PBS containing 1% BSA. Before visualization, the samples were mounted with VECTASHIELD containing DAPI nuclear stain (Vector, Burlingame, CA, USA). Images were captured with a fluorescence microscope (Olympus FSX-100). The optical density of fluorescently stained COL1 was quantified with ImageJ software (NIH, Bethesda, MD, USA) on the basis of the intensity of green staining using a color threshold that included parameters for hue, saturation and brightness.
2.9 Alizarin stainingROBs were cultured in osteogenic differentiation medium, which consisted of DMEM supplemented with 10% FBS, 1% antibiotics, 50 µg/mL ascorbic acid (Sigma Aldrich) and 10 mM β-glycerophosphate (Sigma Aldrich), to promote osteoblast differentiation. Alizarin red staining was performed to determine the presence of extracellular matrix mineralization. After 14 days of culture, the osteoblasts were fixed in 96% ethanol for 15 min and stained with 0.2% alizarin red solution (Sigma Aldrich) in water (pH 6.4) at room temperature for 1 h. Images were captured with a microscope (AXIO Zoom. V16), and the stained area was semiquantified with ImageJ software.
2.9.1 Statistical analysisFor each experiment, three independent experiments for each condition were performed with three or more replicates. The means and standard errors were calculated and analyzed for statistical significance through one-way analysis of variance with Tukey’s test for the live/dead assay, migration assay, COL1 staining, and alizarin red staining, and two-way analysis with the Sidak test was performed for real-time PCR analysis. All analyses were performed with GraphPad Prism 9 software (GraphPad Software, Inc., La Jolla, CA, USA), and * indicates a p value < 0.05, which was considered to indicate statistical significance.
留言 (0)