
記住我
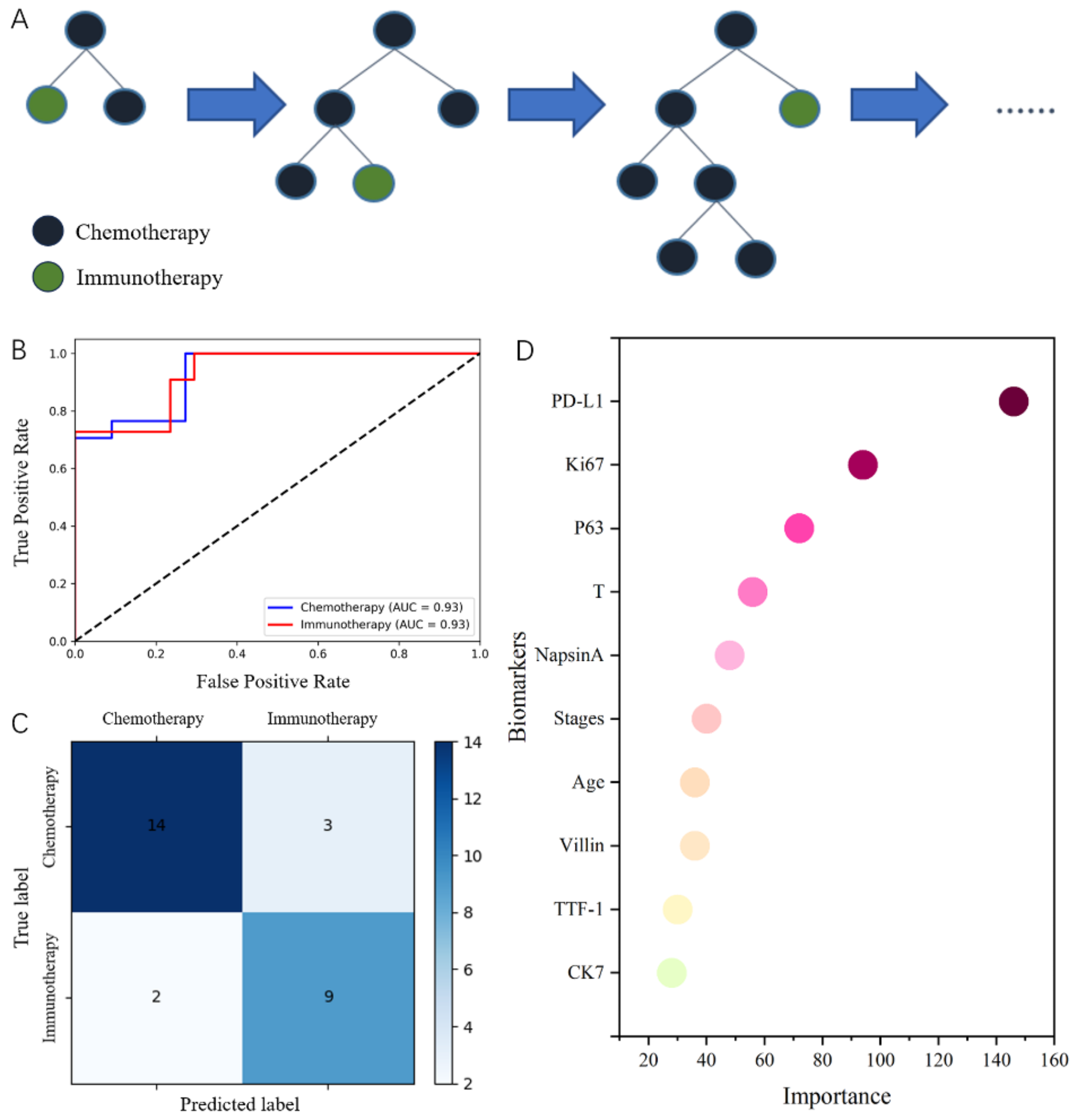
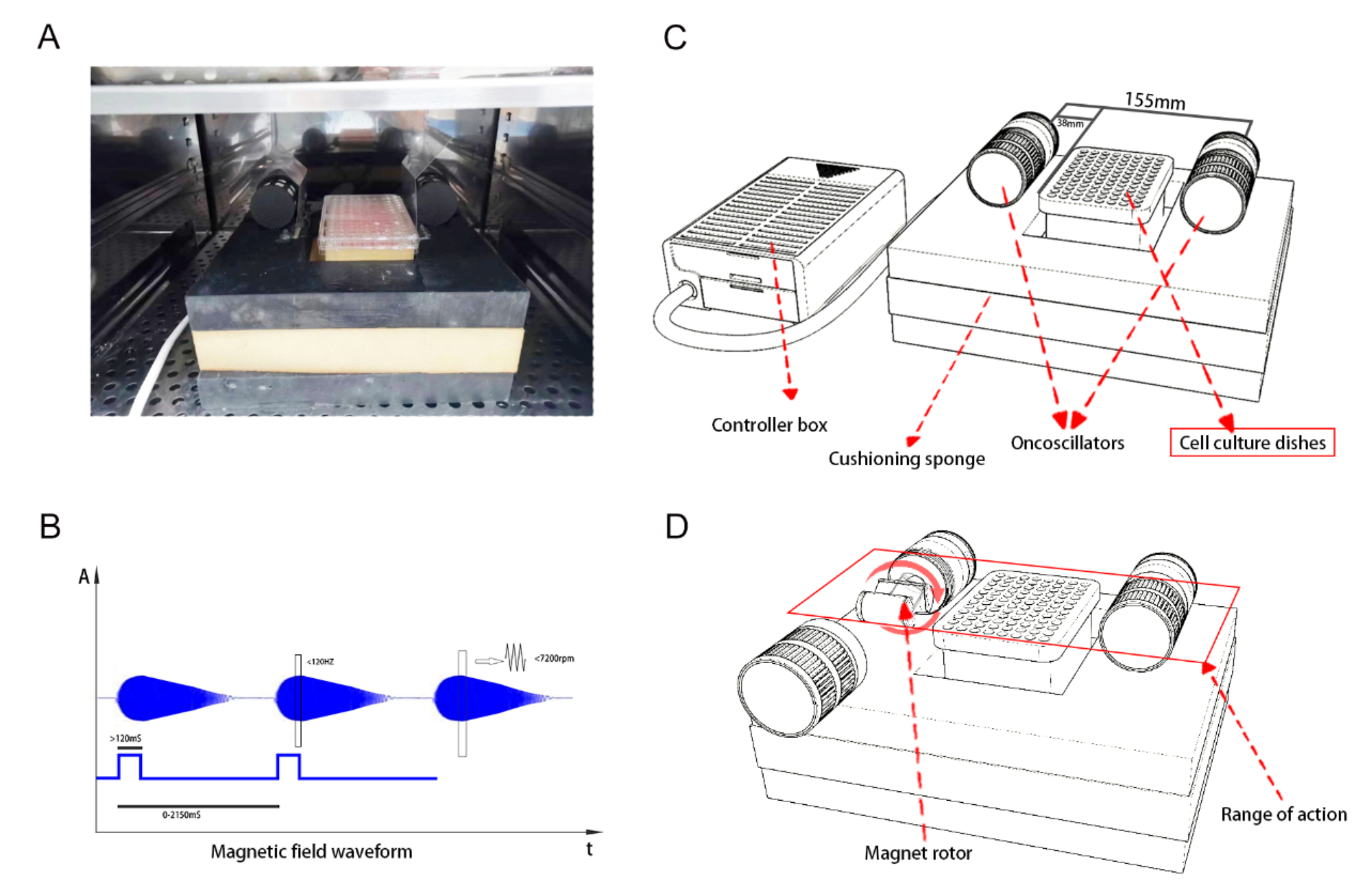

HPV is a highly prevalent sexually transmitted infection (STI) with a significant impact on public health. It is caused by a group of more than 200 related viruses, with about 40 types specifically affecting the genital area as well as the mouth and throat [16, 17]. Despite their prevalence, most HPV infections are asymptomatic and do not lead to serious health problems. However, certain types of HPV are associated with the development of genital warts and various cancers [18]. HPV infection, particularly the HPV16 subtype, is implicated in the etiology of lung and cervical cancers. A key aspect of HPV16 oncogenicity is its ability to increase GLUT1 expression in cancer cells, which is driven by the viral oncoproteins E6 and E7 through the manipulation of various pathways to promote malignancy [19] (Fig. 1).
Fig. 1
HPV and GLUTs Dysregulation. The E6/E7 proteins enhance the nuclear accumulation of HIF-1α through the downregulation of PTEN and its downstream TXNIP. Concurrently, E6/E7 proteins activate the PI3K/AKT pathway, which also results in the nuclear accumulation of HIF-1α. The E6 protein increases the expression of TRX, activating the NF-κB pathway and promoting the transcription of HIF-1α. This nuclear accumulation of HIF-1α upregulates GLUT1 expression at both mRNA and protein levels. The E6 protein also degrades the P53 tumor suppressor protein, leading to the upregulation of GLUT1/4 and an increase in glycolysis through TIGAR. Furthermore, the interaction of E6 with c-Myc upregulates ENO-1, HK1/2, PFK1/2, GLUT1, and LDHA, thereby enhancing glycolysis (E6 does not alter Myc expression levels but increases Myc phosphorylation, leading to enhanced stability and transcriptional activity of c-Myc). * ‘+p’ denotes phosphorylation of the associated protein or molecule
Firstly, the E6 oncoprotein of HPV16 is known to degrade the p53 tumor suppressor protein, thereby inhibiting apoptosis and altering glucose metabolism. This degradation leads to increased glycolysis through Tp53-induced glycolysis and apoptosis regulator (TIGAR) and the upregulation of GLUT1 and GLUT4, which is a hallmark of cancer cell metabolism [9, 20, 21].
Additionally, c-Myc, a well-known oncogene, interacts with E6, resulting in the upregulation of glycolysis-related enzymes, including enolase 1 (ENO-1), hexokinase 1/2 (HK1/2) phosphofructokinase 1/2 (PFK1/2), GLUT1, and lactate dehydrogenase A (LDHA). This interaction underscores the complex network of oncogenic signals that promote cancer cell survival and proliferation [22,23,24].
In lung cancer cell lines, the overexpression of E6 has been shown to increase the expression of thioredoxin (Trx), a disulfide reductase that plays a role in the intracellular antioxidant system and is necessary for maintaining oxidative stress balance and protecting cells from oxidative damage. Trx, in turn, activates the NF-κB pathway, a transcription factor that results in the transcription of hypoxia-inducible factor − 1 α (HIF-1α), which upregulates GLUT1 expression. Additionally, the activation of NF-κB results in the nuclear translocation of P65 and, finally, upregulation of GLUT1 mRNA and protein levels [25].
The thioredoxin-interacting protein (TXNIP) regulates the intracellular redox state and glucose metabolism, which are associated with GLUT1 expression and glucose uptake. In lung cancer, HPV16 E6/E7 proteins downregulate PTEN, leading to the inhibition of TXNIP expression. This inhibition results in the accumulation of HIF-1α by inhibiting the translocation of nuclear HIF-1α to the cytoplasm and the upregulation of GLUT1 expression at both mRNA and protein levels [26].
Furthermore, HPV infection, particularly with high-risk types like HPV-16, significantly alters cellular signaling pathways, including the PI3K/Akt/HIF-1α/GLUT axis. The HPV oncoproteins E6 and E7 activate the PI3K/Akt signaling pathway, leading to several downstream effects crucial for cancer progression, including metabolic changes. The activation of the PI3K/Akt pathway can induce the stabilization and activation of HIF-1α, even under normoxic conditions. This HPV-induced pseudo-hypoxic state promotes the expression of genes involved in angiogenesis, metabolism, and epithelial-mesenchymal transition (EMT). HIF-1α activation, in turn, increases the expression of various GLUTs, particularly GLUT1 [27]. When investigating the prognostic value of cervical cancer, high GLUT1 expression and the presence of the HPV16 subtype are independent prognostic factors for overall survival. High GLUT1 expression is associated with older age, squamous cell carcinoma, advanced tumor stage, pelvic lymph node metastases, and a lower rate of hysterectomy. The prognostic impact is particularly significant in the HPV16-positive group, which also exhibits decreased immune cell scores [28]. Furthermore, another study found that cervical tumor cells enhance glucose utilization by upregulating GLUT1. According to the strong induction of GLUT1 mRNA and protein in HPV-positive cervical squamous intraepithelial neoplasia 3 (CIN 3) lesions, it was concluded that GLUT1 overexpression is an early event in cervical neoplasia [29].
In summary, the HPV16 subtype is a critical factor in the development of lung and cervical cancers, primarily through the modulation of glucose metabolism by its E6 and E7 oncoproteins. The upregulation of GLUT1 expression is a key event in this process, with significant implications for cancer prognosis and potential therapeutic strategies.
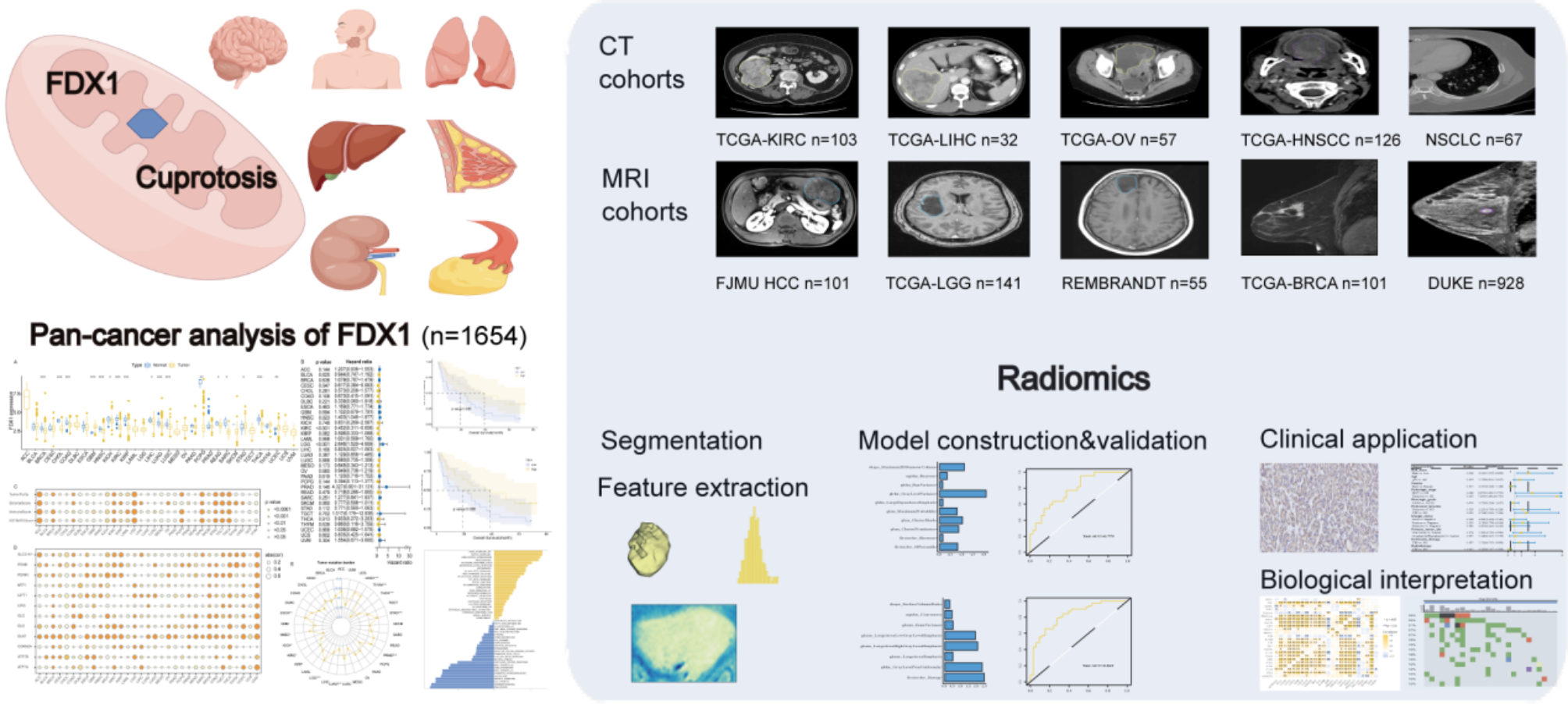
HCVHCV is a single-stranded RNA virus that belongs to the Flaviviridae family. Within infected individuals, the RNA replication process of HCV is error-prone, resulting in high genetic diversity and the emergence of genetically related variants called quasispecies. Variations in the hypervariable region can indicate the presence of different quasispecies, which may contribute to the chronicity of HCV infection. The genome of the virus consists of a single open reading frame (ORF), which is made up of 3010 amino acids. This ORF is flanked by non-translated regions (NTRs) at the 5’ and 3’ ends, which play a crucial role in viral replication and translation. The 5’ NTR contains an internal ribosome entry site that is responsible for initiating the translation of the HCV protein. The translation of the HCV protein leads to the formation of a single polyprotein. This polyprotein is then processed by cellular proteases to produce the structural proteins that make up the viral particles, such as the core and envelope glycoproteins E1 and E2. The polyprotein is alternatively processed by viral proteases to produce nonstructural (NS) proteins such as p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B. As of 2015, an estimated 71 million people worldwide were affected by chronic HCV infection, with varying prevalence rates across regions. The primary modes of HCV transmission include transfusion of blood products and unsafe healthcare practices [30, 31]. HCV infection is a major cause of advanced hepatic fibrosis and cirrhosis, with a significantly increased risk for the development of hepatocellular carcinoma (HCC) [32] (Fig. 2).
Fig. 2
HCV and GLUTs Dysregulation. The HCV core protein leads to the upregulation of TNF-α and interleukin-6, and the downregulation of the TSC1/TSC2 complex. This combination results in the degradation of IRS1/2, leading to the downregulation of the PI3K/AKT pathway. This downregulation subsequently decreases the translocation of GLUT4 to the cellular membrane and increases the nuclear accumulation of FoxO1. The HCV non-structural proteins NS4A and NS5A induce. mitochondrial damage, leading to the production of reactive oxygen species (ROS) and activation of the JNK pathway. This results in decreased phosphorylation of FoxO1, leading to its nuclear accumulation. FoxO1 is a regulatory protein involved in glucose metabolism. The nuclear accumulation of FoxO1 upregulates gene expression of PEPCK and G6Pase, leading to increased gluconeogenesis and glucose production, and the downregulation of GLUT2/4.* ‘+p’ denotes phosphorylation of the associated protein or molecule
HCV infection disrupts glucose metabolism through various mechanisms. Notably, the HCV proteins NS4A and NS5A induce mitochondrial damage, leading to the production of reactive oxygen species and activation of the JNK pathway. This ultimately results in decreased phosphorylation and nuclear accumulation of FoxO1, a regulatory protein involved in glucose metabolism of the liver, muscle, adipose tissue, and pancreas, thereby impacting glucose metabolism by upregulating enzymes involved in glucose production pathways, such as glucose-6 phosphatase and phosphoenolpyruvate carboxy kinase 2, which result in increased gluconeogenesis, glycolysis, and down-regulation of GLUT2/4 [33, 34]. The downregulation of GLUT2/4 in HCV infection, while seemingly counterintuitive, aligns with observations in overall hepatocellular carcinoma (HCC). In HCC, GLUT2 expression often decreases in preneoplastic and neoplastic hepatic lesions, while other GLUTs, particularly GLUT1, may be upregulated.
Furthermore, HCV core proteins interfere with insulin signaling pathways by upregulating the Ser312 phosphorylation of insulin receptor substrate 1 (IRS-1) (the key molecule involved in the propagation of insulin signal downstream from the insulin receptor), promoting its degradation and impairing insulin signaling to the PI3K/Akt pathway. Additionally, HCV proteins interact with cellular structures such as the endoplasmic reticulum (ER) and mitochondria, resulting in oxidative stress and the upregulation of pro-inflammatory cytokines like tumor necrosis factor-α (TNF-α), interleukin 8, interleukin 6 (IL-6), transforming growth factor β (TGF-β), suppressor of cytokines (SOC3), and Fas ligand. This infection also down-regulates the TSC1/TSC2 complex and causes subsequent upregulation of mTOR/S6K1, which leads to Ser1101 phosphorylation of IRS-1 and its subsequent degradation. These events, along with the down-regulation of the TSC1/TSC2 complex and the induction of TNF-α/IL-6 production, disrupt insulin-mediated transactivation of the glucose transporter GLUT4, leading to reduced glucose uptake and contribute to insulin resistance and tumorigenesis [35,36,37].
In HCC, various glucose transporters, including GLUT1, GLUT2, GLUT3, GLUT5, GLUT6, and GLUT9, play complex roles. High expression of GLUT1 is associated with larger tumor size, increased vascular invasion, and a poorer prognosis. GLUT2 expression is elevated in HCC but decreased in preneoplastic and neoplastic hepatic lesions. High expression of GLUT3 is linked to elevated α-fetoprotein levels, large tumor size, poor histological differentiation, poor prognosis, and reduced overall survival in HCC [38,39,40]. Other GLUTs, such as GLUT5, GLUT6, and GLUT9, also exhibit significant involvement in HCC. For example, GLUT5 expression is significantly higher in liver metastases compared to primary lung tumors and is elevated in liver carcinoma [9, 41]. These findings highlight the diverse roles of different GLUTs in HCC and suggest potential implications for future research and therapeutic strategies.
It is important to note that studies specifically discussing GLUT status in HCV-associated HCC are scarce. In this review, we have discussed GLUT regulation in HCV infection and HCC separately, considering the overlapping pathways between HCV infection and HCC development as potentially sharing similar etiologies. While HCV infection influences glucose metabolism through modulation of GLUT1, GLUT2, and GLUT4, the alterations in GLUT expression in HCC are not necessarily specific to HCV-related cases. For instance, the overexpression of various GLUTs, including GLUT1, GLUT2, GLUT3, GLUT5, GLUT6, and GLUT9, has been observed in HCC regardless of etiology. Future research should focus on elucidating the specific mechanisms by which HCV infection may contribute to altered GLUT expression in the context of HCC development.
In summary, HCV infection influences glucose metabolism through the modulation of GLUT1, GLUT2, and GLUT4, contributing to the pathogenesis of the diseases associated with this virus.
HTLVHTLV-1 is the first human oncogenic retrovirus that has been identified as the causative agent of two severe diseases: adult T cell leukemia/lymphoma and myelopathy/tropical spastic paraparesis [42, 43]. HTLV-1 infects nearly 20 million individuals worldwide, with Japan, Africa, the Caribbean, and South America as the endemic regions [44]. The mechanisms of HTLV-1 infection and its cellular tropism are complex and not fully elucidated. Several receptors are important in HTLV-1 transmission. For instance, HTLV-1 attaches to heparan sulfate proteoglycan, which is present in the host cell. Additionally, the envelope proteins of the virus, particularly their interactions with the GLUT1 receptor, play a pivotal role in the virus’s attachment, entry, and subsequent pathogenicity in host cells [45,46,47,48]. The virus’s life cycle and pathogenicity are influenced by its regulatory and accessory genes, with the regulatory gene Tax-1 playing a crucial role in virus replication and the transcription of viral gene products. The PDZ binding motif at the carboxyl terminus of Tax-1 is important in cellular transformation. GLUT1 is a key receptor for HTLV-1, and its interaction with the virus’s envelope glycoproteins is essential for the virus’s entry into host cells. The expression of GLUT1 is not inherent in quiescent primary T cells but is induced upon T cell activation, which is significant as it enhances the infectivity of HTLV-1. Moreover, the expression of GLUT1 on T lymphocytes is increased by mitogens or TGF-β, which in turn increases the infectivity of HTLV-I [49]. Interestingly, while overexpression of GLUT1 in cells that produce HTLV-1 virus-like particles reduces infectivity, overexpression of GLUT3, a closely related protein, does not have the same effect. This specificity suggests that GLUT1’s role in HTLV-1 pathogenesis is unique and not easily replicated by other glucose transporters [47] (Fig. 3).
TAX-1 is a multifunctional protein that not only drives the transcription of viral gene products but also interacts with various host cell proteins to modulate the virus’s life cycle. One such interaction is with nexin 27 (SNX27), which is involved in the recycling of GLUT1 from endosomal vesicles to the plasma membrane. This interaction is crucial for maintaining GLUT1 expression on the cell surface, and disruption of this interaction leads to reduced surface expression of GLUT1, increased release of HTLV-1 into the supernatant, and decreased cell-to-cell-mediated HTLV-1 infection. The interaction between TAX-1 and SNX27 may alter this regulation, potentially affecting the receptor molecule’s availability for HTLV-1. Moreover, plasma membrane-bound receptor molecules have been shown to interfere with virion release and infectivity in other retroviruses, including human immunodeficiency virus (HIV) [50].
TGF-β has an important role in preventing the growth of T cells during the mid-G1 phase as well as in promoting tumorigenesis. It achieves this by binding to the Type I (TβRI) and Type II (TβRII) serine/threonine kinase receptor complex, which triggers the phosphorylation of downstream targets, including the Smad proteins. The resulting Smad2/3/4 complex then moves to the nucleus, leading to the increased expression of GLUT1. However, the protein TAX-1 can inhibit this Smad-dependent pathway, which results in a decrease in GLUT1 expression [51].
Overexpression of GLUT1 in HTLV-1-resistant cell lines, such as MDBK, increases the titer of HTLV-1 and HTLV-2 Env-pseudotyped particles [52]. This finding indicates that the level of GLUT1 in target cells correlates with the titer of the HTLV-1/2 Env-pseudotyped virus, suggesting that GLUT1 availability is a limiting factor for HTLV-1 infectivity [53].
In summary, the interactions between HTLV-1, GLUT1, and SNX27 are critical for the virus’s life cycle and pathogenesis. The protein interaction between TAX-1 and SNX27 play a significant role in modulating the localization and expression of GLUT1, which in turn affects the infectivity and transmission of HTLV-1. Understanding these molecular mechanisms provides insight into potential therapeutic targets for the treatment of HTLV-1-associated diseases.
Fig. 3
HTLV1 and GLUTs Dysregulation. TGF-β promotes the phosphorylation of Smad proteins. The phosphorylated Smad 2/3/4 complex translocates to the nucleus, leading to increased expression of GLUT1. The SNX27 protein promotes the recycling of GLUT1 to the plasma membrane. TAX-1 downregulates the Smad and SNX27 pathways, resulting in decreased expression and translocation of GLUT1 * ‘+p’ denotes phosphorylation of the associated protein or molecule. ** ‘p’ vector indicates phosphorylated proteins
EBVEBV, also known as human herpesvirus 4, is a gamma herpesvirus that infects a large portion of the human population. While generally harmless, EBV can cause infectious mononucleosis, commonly known as mono, in teenagers or young adults. In rare cases, individuals with a healthy immune system can experience chronic active infection [54]. However, EBV has also been linked to various types of cancers, including Burkitt’s lymphoma, Hodgkin’s lymphoma, natural killer cell lymphoma, nasopharyngeal carcinoma, post-transplant lymphoproliferative disorder, and EBV-positive gastric adenocarcinoma [55]. EBV induces extensive methylation in both the host and viral genomes, which plays a role in facilitating cellular functions that support virus persistence and propagation. Tumors that are positive for EBV exhibit distinct genetic alterations compared to EBV-negative tumors. For example, EBV-positive gastric adenocarcinoma often shows recurring PIK3CA mutations, extensive DNA hypermethylation, and amplification of the JAK2, CD274, and PDCD1LG2 genes [56].
Nasopharyngeal carcinoma (NPC) is a type of cancer that is strongly associated with EBV. NPC cells tend to have increased expression of GLUT1. This increased expression of GLUT1 is associated with lymph node metastasis and clinical stage [57]. The Epstein-Barr virus latent membrane protein 1 (LMP1) plays a critical role in the transformation of B-cells by mediating NF-κB and AKT/mTORC1 signaling pathways, which in turn influence the expression of GLUT1. As a result, GLUT1, which is typically expressed at low levels, becomes highly expressed during tumorigenesis and is considered a marker of tumor progression. LMP1 upregulates the transcriptional mRNA and protein levels of GLUT1, and knocking down LMP1 expression significantly reduces glucose uptake. Independent data also confirmed these results in different types of EBV-related cancer, such as B-cell lymphomas [58, 59].
Additionally, LMP1 contributes to metabolic reprogramming by disrupting the regulation of glycolysis enzymes such as hexokinase 2 [60]. This reprogramming of EBV-mediated glycolysis in NPC cells leads to increased production of IL-1β, IL-6, and GM-CSF, which results in the induction of myeloid-derived suppressor cells that contribute to T-cell suppression. Similar dysregulation of GLUT1 by LMP1-activated NF-κB has been observed in other types of EBV-related cancers, such as B-cell lymphomas [61].
In summary, EBV, particularly LMP1, influences glucose metabolism in cancer cells, including the upregulation of GLUT1, through the activation of NF-κB and mTORC1 signaling pathways. This alteration in glucose metabolism is a hallmark of various types of cancer, including NPC associated with EBV infection. GLUT1 overexpression in NPC is correlated with tumor progression and metastasis (Fig. 4).
Fig. 4
EBV and GLUTs Dysregulation. LMP1 upregulates the AKT/mTORC1 and NF-κB pathways. Additionally, the activation of AKT/mTORC1 modulates NF-κB. This activation of NF-κB upregulates the expression of GLUT1 through the P50 and P65 heterodimers. The upregulation of GLUT1 and deregulation of HK2 lead to increased production of IL-1β, IL-6, and GM-CSF, resulting in the induction of myeloid-derived suppressor cells (MDSCs). * ‘+p’ denotes phosphorylation of the associated protein or molecule
HBVHBV is a DNA virus that specifically infects hepatocytes (liver cells) and is a leading cause of liver cirrhosis and hepatocellular carcinoma worldwide [62, 63]. The virus enters hepatocytes through the binding of the large HBV surface proteins to the HBV receptor on the cell surface, known as sodium taurocholate co-transporting polypeptide. In chronic HBV carriers, the accumulation of the HBV large surface proteins (LHBs) can trigger an ER overload response, leading to ER stress-mediated cell proliferation, metabolic changes, and genomic instability [64].
One significant consequence of chronic HBV infection is metabolic alterations observed in patients, particularly during the immune-tolerance phase [65]. ER stress signaling, specifically XBP1 (a major, well-conserved component of the unfolded protein response (UPR)), plays a crucial role in regulating glucose metabolism through pathways involving hypoxia-inducible factor 1 (HIF1), GLUT1, and GLUT2. The HIF1 pathway is activated under conditions of low oxygen levels (hypoxia) and plays a vital role in cellular adaptation to hypoxic environments [66, 67]. In the context of HBV infection, HIF1 signaling can induce the transcription of GLUT1 as well as several rate-limiting glycolytic enzymes. This adaptive response may allow hepatocytes to overcome the energy imbalance caused by the hypoxia-induced decline in mitochondrial activity [68, 69] (Fig. 5).
In recent years, many studies have demonstrated that HBV replication depends on the AMPK-ULK1-induced autophagy and Akt/mTOR suppressed ULK1-induced autophagy pathways. Low glucose levels and inhibition of GLUTs activate the AMPK-mTOR-ULK1-autophagy axis in hepatocytes, which is involved in HBV replication [68,69,70,71]. Manipulating glucose concentrations and using inhibitors like 2-DG, which inhibit glycolysis and activate the Akt/mTOR pathway, can suppress HBV replication [72].
In summary, HBV infection influences glucose metabolism through the modulation of GLUT1 and GLUT2, contributing to the pathogenesis of the diseases associated with this virus. This understanding of the role of GLUT in HBV infection could provide valuable insights for future research and potential therapeutic strategies.
Fig. 5
HBV and GLUTs Dysregulation. HBV replication, in conjunction with low glucose concentration and hypoxia, induces ER stress, which in turn triggers the splicing of XBP1 to its activated form. XBP1 enhances the expression of HIF-1α, which is further stabilized from degradation under hypoxic conditions. XBP1 cooperates with HIF-1α to increase the expression of GLUT1. Additionally, the low glucose concentration in the infected cells augments HBV replication via the AKT/mTORC1/ULK1 and AMPK/ULK1 pathways. Additionally, hypoxia inhibits HIF-1 degradation, leading to HIF-1 nuclear accumulation *UPR: The Unfolded Protein Response
KSHVKSHV, also known as human herpesvirus 8, is a virus that can cause several human malignancies, primarily in immunocompromised individuals. These malignancies include Kaposi’s sarcoma (KS), primary effusion lymphoma, and multicentric Castleman’s disease. KSHV can establish a persistent infection by becoming latent in CD19+ peripheral B-lymphocytes, with the viral genome persisting as a circular episome in the host cell’s nucleus. KSHV exhibits a biphasic life cycle consisting of a latent phase and a transient lytic reactivation phase. During the latent phase, the latency-associated nuclear antigen plays a crucial role in maintaining latency and ensuring proper distribution of viral episomes during cell division [73,74,75]. It is important to note that while KSHV can establish a persistent infection in CD19+ peripheral B-lymphocytes, KS, one of the main malignancies associated with KSHV, is of endothelial cell origin. The prevalence of KSHV varies geographically, with higher rates observed in Sub-Saharan Africa and the Mediterranean region. KSHV infection is most commonly associated with immunodeficiency states, including HIV infection, iatrogenic immunodeficiency, and aging [76] (Fig. 6).
KSHV-infected cells have been found to influence various signal transduction pathways, including the PI3K/Akt/mTOR signaling pathway. This pathway plays a vital role in regulating multiple biological processes, including apoptosis, metabolism, cell proliferation, and cell growth. By manipulating this pathway, KSHV gains an advantage in promoting the growth and survival of infected cells, particularly those with high proliferation rates that rely on glucose metabolism [76, 77]. PTEN, a dual protein/lipid phosphatase, inhibits the PI3K/Akt pathway primarily by targeting phosphatidylinositol 3,4,5-triphosphate produced by PI3K. In fact, PTEN acts as a tumor suppressor by counteracting the activation of the PI3K/Akt pathway and inhibiting cell growth and proliferation [78].
The PI3K/Akt/mTOR pathway is involved in regulating glucose metabolism, including glucose uptake and glycolysis. Activation of this pathway promotes the expression of GLUT1 and re-localizes it to the cell membrane, thereby increasing glucose uptake [79]. This alteration in GLUT1 expression and membrane exposure is closely linked to cancer progression, as cancer cells require efficient biosynthesis of macromolecules to support their high proliferation rates. The altered GLUT1 membrane trafficking in KSHV-infected cells increases their sensitivity to cell death induced by the glycolysis inhibitor 2-Deoxy-D-glucose (2DG), which inhibits glycolysis [
留言 (0)