
記住我




Myelofibrosis (MF) stands as an intricate and challenging entity within the expansive domain of BCR::ABL1 negative myeloproliferative neoplasms (MPNs) [1]. It manifests as a disorder characterized by the aberrant proliferation of hematopoietic stem cells, precipitating a deregulated cascade in the production of blood cells [2]. Having been initially acknowledged over a century ago in 1879 with Gustav Heuck [3], MF has been the subject of exhaustive research endeavors that have substantially augmented our comprehension of its pathophysiological underpinnings, clinical phenotypes, and therapeutic modalities. This complex pathology not only poses formidable clinical challenges but also serves as an archetypal model for investigating the sophisticated interplay between genetic determinants, the bone marrow (BM) microenvironment, intricate interactions with visceral organs such as the spleen, and the orchestrating influence of the immune system.
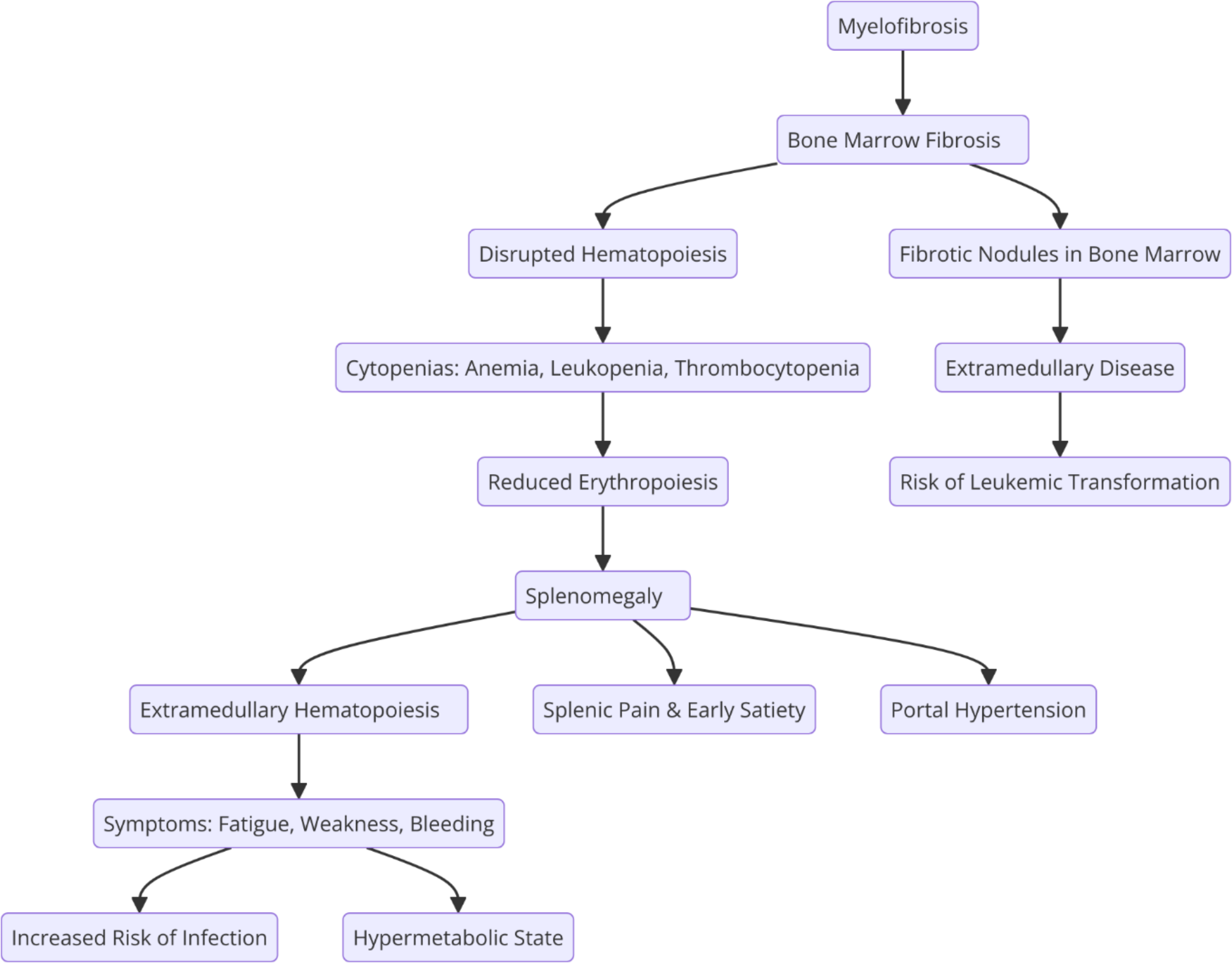
Central to the pathology of MF is the distinctive development of fibrous tissue within the BM milieu, compromising the physiological architecture imperative for hematopoiesis. This fibrotic metamorphosis precipitates a myriad of clinical complications ranging from anemia and splenomegaly to an augmented propensity for leukemic transformation [4] as shown in the following diagram.
Fig. 1
Pathophysiological relationship between MF, BM fibrosis, and splenomegaly. The diagram illustrates the progression from BM fibrosis leading to disrupted hematopoiesis, compensatory splenic involvement with extramedullary hematopoiesis, and the subsequent clinical manifestations, including cytopenias, splenic enlargement, and increased risk of leukemic transformation
This disorder is dichotomously classified into Primary Myelofibrosis (PMF), originating de novo without antecedent myeloproliferative conditions, and Secondary Myelofibrosis, evolving as a sequel from prior myeloproliferative disorders such as Polycythemia Vera or Essential Thrombocythemia [1, 5].
Predominant among the identified genetic mutations instigating MF are those affecting JAK2, CALR, and MPL genes [6]. These driver mutations activate signaling pathways pivotal to hematopoiesis, consequently fostering unbridled proliferation of hematopoietic stem and progenitor cells. The JAK-STAT pathway, particularly accentuated by the landmark discovery of the JAK2V617F mutation [7], represents a watershed moment in comprehending the molecular substrates of MPNs, including MF.
To note that a spectrum of additional non-driver genetic mutations imparts considerable features to the disease’s progression and prognosis, for instance genes implicated in splicing factor ZRSR2, SRSF2, SF3B1 [8], epigenetic DNMT3A, TET2, metabolic IDH1, IDH2 [9], and other pathways TP53, KRAS, NRAS [10, 11], . Furthermore, the dynamic interplay between hematopoietic cells and the BM microenvironment, consisting of stromal cells, extracellular matrix (ECM), and a milieu of cytokines, emerges as a pivotal factor in MF pathogenesis. Aberrant interactions contribute significantly to the fibrotic transformation. Noteworthy cytokines implicated in BM homeostasis deregulation encompass Transforming Growth Factor-beta (TGF-β), Fibroblast Growth Factor (FGF) [12], and proinflammatory cytokines like Tumor Necrosis Factor-alpha (TNF-α) [13].
Recent research has significantly expanded our understanding of how non-driver mutations influence the progression and transformation of MF, particularly PMF. Non-driver mutations, such as those in ASXL1, EZH2, TET2, SRSF2, and IDH1/IDH2, have been found to exacerbate disease severity, accelerate progression, and increase the risk of transformation to acute myeloid leukemia (AML).
These studies have shown that mutations in genes like ASXL1 and EZH2 are linked to faster disease progression, higher degrees of BM fibrosis, and shorter survival rates. These mutations tend to drive clonal evolution, contributing to genetic complexity and heterogeneity in the disease, which worsens the overall prognosis [14, 15]. ASXL1 mutations, for instance, are associated with a more aggressive disease course, leading to poor response to therapies such as JAK inhibitors and a higher likelihood of leukemic transformation [16]. Moreover, mutations in SRSF2 and IDH1/IDH2 have been linked to impaired cell differentiation and increased myeloproliferation, further exacerbating the risk of transition to AML [17]. The presence of EZH2 mutations has been found to disrupt epigenetic regulation, accelerating both fibrosis and leukemic transformation, often marking patients for worse outcomes [15].
The accumulation of non-driver mutations also directly influences treatment response. Patients harboring mutations in genes such as ASXL1, SRSF2, and U2AF1 often show resistance to JAK inhibitors like Ruxolitinib, requiring alternative therapeutic strategies, such as combination therapies targeting epigenetic or splicing mechanisms [15]. Importantly, the number of mutations also plays a role: a higher mutational burden, particularly involving multiple high-risk mutations, is associated with increased chances of disease progression and leukemic transformation [17]. This makes comprehensive genetic testing crucial for risk stratification, as identifying these mutations can guide personalized treatment approaches aimed at mitigating disease progression and improving patient outcomes. A comprehensive study conducted across 60 institutions in Spain analyzed the impact of these mutations in 312 patients with PMF using targeted next-generation sequencing (NGS). This study revealed that 72% of patients had non-driver mutations, with 47% harboring two or more mutations. The most frequent mutations detected were in ASXL1 (34%), TET2 (22%), SRSF2 (17%), U2AF1 Q157 (9%), CBL (8%), EZH2 (7%), TP53 (6%), DNMT3A (6%), and SETBP1 (5%). The presence of these mutations was strongly associated with worse overall survival (OS). Specifically, patients without mutations had a median OS of 12.4 years, whereas those with one mutation had a median OS of 8.7 years, and those with two or more mutations had a significantly reduced median OS of 4.4 years (P < .001) [18].
These findings underscore the adverse prognostic impact of non-driver mutations, which contribute to poorer clinical outcomes and higher mortality rates. Notably, ASXL1 mutations emerged as the most prevalent high-risk mutations, with a variant allele frequency > 20% identified as a critical adverse prognostic factor, particularly influencing decisions regarding allogeneic hematopoietic stem cell transplantation (allo-HSCT). The study also highlighted the independent prognostic significance of TP53, CBL, and SETBP1 mutations, advocating for their inclusion in the Molecular International Prognostic Scoring System (MIPSS70 and MIPSS70 plus) models for PMF. This aligns with previous findings suggesting that mutations in these genes should be considered in prognostic models to better stratify patients and tailor treatment strategies [19, 20]. Overall, the integration of non-driver mutations into clinical practice is crucial for improving risk stratification and guiding personalized treatment approaches in MF. These mutations not only exacerbate the disease course but also complicate treatment, often leading to resistance to therapies like JAK inhibitors, and accelerating progression to more severe stages, including AML [14,15,16].
MF, embodying a formidable and multifaceted clinical entity, epitomizes the intricate symbiosis between genetic mutations, deregulated signaling pathways and aberrant interactions between hematopoietic and stromal cells within the BM microenvironment and extramedullary niches [21]. The evolving comprehension of its molecular foundations has smoothed the path for targeted therapeutic modalities, instilling optimism for enhanced disease management [22]. Nevertheless, the intricate nature of MF continues to propel ongoing research endeavors, aiming to unravel deeper insights into its pathogenesis and foster innovative therapeutic strategies to effectively address the diverse clinical challenges posed by this malady.
The clinical manifestations of MF exhibit a spectrum of diversity, spanning from mild constitutional symptoms to severe, life-threatening complications [23]. Commonly encountered symptoms include anemia, myeloblastosis, fatigue, and weight loss, reflective of compromised normal blood cell production (Fig. 1) [24]. Organomegaly, including splenomegaly and hepatomegaly, indicative of the pervasive impact on visceral organs, is a hallmark feature that results from extramedullary hematopoiesis (EMH) and contributes to abdominal discomfort and early satiety [25]. Simultaneously, the progressive fibrosis within the BM and deviations in blood cell counts manifest as anemia, thrombocytopenia, and leukopenia, further exacerbated by cytopenia-associated bone pain and fatigue necessitating interventions such as blood transfusions or other supportive measures [26, 27].
As we pore over the intricate molecular landscape, a comprehensive understanding of these classifications, clinical features and genetic aberrations becomes pivotal for advancing diagnostic precision and therapeutic interventions in the realm of MF. The ongoing quest for unraveling the complex facets of MF underscores the imperative to refine our knowledge base, ultimately enhancing diagnostic accuracy and facilitating targeted therapeutic innovations for improved patient outcomes.
Bone marrow niche and microenvironmentThe BM niche constitutes a specialized microenvironment crucial for the maintenance and regulation of hematopoietic stem cells (HSCs). Comprising stromal cells, endothelial cells, osteoblasts, and ECM, the niche orchestrates the delicate balance between HSCs self-renewal and differentiation [28]. In MF, this equilibrium is disrupted, precipitating aberrant cellular behaviors within the BM microenvironment. Abnormal signaling from mutated hematopoietic cells prompts the proliferation of fibroblasts, culminating in heightened collagen deposition, characteristic of fibrosis development. Furthermore, somatic mutations in JAK2, CALR, and MPL disturb signaling pathways within the niche, further perturbing the intricate dance between HSCs fate decisions [29]. Particularly, the persistent JAK-STAT signaling promote the uncontrolled expansion of myeloid progenitors.
As it is known, the hallmark of MF is BM fibrosis, characterized by the deposition of fibrous tissue that disturbs normal hematopoietic architecture. Activated stromal cells, including fibroblasts and myofibroblasts, contribute to excessive production and deposition of ECM proteins, notably collagen. The upregulation of TGF-β further stimulates fibroblast activation [30]. Important to mention alterations in the interactions between hematopoietic cells and the ECM that contribute to the fibrotic process. Actually, changes in the ECM’s composition and organization affect normal homing, retention, and differentiation of HSPCs [31].
Furthermore, the inflammatory microenvironment emerges as a distinguishing feature of MF, marked by the deregulated production of cytokines and chemokines affecting both hematopoietic and stromal cells [29, 32]. Immune deregulation within the microenvironment, with abnormal activation of immune cells, may contribute to disease pathogenesis and progression. In statement inflammatory cytokines, such as TNF-α, IL-6, and IL-1, are elevated in MF, disrupting the BM microenvironment [33].
These cytokines promote the vicious circle where stimulating fibrotic factor production, enhance mutant cell survival, and contribute to a pro-inflammatory milieu that sustains the disease.
Epigenetic changes, particularly altered DNA methylation patterns, further compound the complexity, influencing gene expression dynamics in both hematopoietic and stromal cells [34].
MF is linked to angiogenic changes in the BM vasculature, causing disruptions in normal vascular architecture and contributing to hypoxia. Key regulators of angiogenesis, including angiopoietin-1 and angiopoietin-2, are deregulated [13].
For these reasons MF involves a complex interplay of genetic mutations, deregulated signaling pathways, and changes in the BM microenvironment. Unraveling these intricacies is essential for developing effective interventions that address the root causes and improve outcomes for individuals affected by MF.
Role of the spleen in myelofibrosisSplenomegaly is a prominent feature in the landscape of MF, driven by numerous alterations in the hematopoietic system [35]. Excessive EMH, typically confined to the BM, extends to the spleen due to the perturbed BM microenvironment [36]. This expansion is further accentuated by the spleen’s pivotal role as a reservoir for blood cells, especially red blood cells, compensating for compromised production in the altered BM. Pathological changes within the spleen mirror those observed in the BM, featuring the development of fibrosis and consequential alterations in the microenvironment, potentially impairing normal splenic functions [5].
The spleen plays a crucial role in the pathophysiology of MF, acting as a reservoir for malignant HSCs that retain a full differentiation program. This suggests that the spleen is not only a site of EMH but also significantly contributes to the maintenance and progression of the disease. The interaction between these malignant HSCs and the splenic microenvironment, particularly mesenchymal stromal cells (MSCs), is key to regulating this aberrant hematopoiesis. MSCs in the spleen provide a supportive niche that enables the survival and proliferation of these malignant cells, thereby promoting disease progression [37, 38].
Our preliminary data, presented at the EHA Congress 2024, further elucidate the complex molecular landscape of the spleen in MF patients. Utilizing spatial transcriptomic analyses on spleen specimens from MF patients, we observed a modulated gradient of gene expression across distinct spleen regions. Specifically, there is a decremental gradient in gene expression related to inflammation and ECM remodeling from intravascular to perivascular regions, culminating in the red pulp. Conversely, an incremental expression gradient is noted in genes associated with iron metabolism, hemoglobin synthesis, oxygen transport, and erythropoiesis [39]. These findings suggest a dynamic orchestration of inflammatory responses, ECM alterations, and EMH within the spleen, characterizing MF and we can mint the concept of the spleen as a “tumor surrogate”. In fact in MF the spleen encapsulates its transformation into a site of massive infiltration by abnormal hematopoietic cells, especially in advanced disease stages (Fig. 2). While metaphorically labelled a “tumor surrogate”, the spleen in MF does not adhere to the characteristics of a true neoplastic mass. Instead, this term underscores the invasive and disruptive nature of the spleen’s altered state, akin to a pathological mass, albeit without the hallmark features of a malignancy [37,38,39].
The findings from our preliminary study underscore the need for further investigation into targeted therapies that address the unique spatial characteristics of the splenic microenvironment in MF.
Moreover spleen volume, often used as a surrogate marker for disease response in MF, reflects not only the disease burden but also serves as an indirect measure of tumor activity and the splenic microenvironment’s role in sustaining the malignant clone. Changes in spleen size could directly relate to shifts in the underlying disease biology, making it a crucial parameter in both prognosis and treatment decisions [40, 41]. These integrated insights highlight the spleen’s dual role in sustaining the disease and serving as a measurable indicator of therapeutic efficacy in MF, making it a focal point in both understanding and managing the disease.
Clinically, the consequences of splenomegaly in MF extend beyond mere organ enlargement. Symptoms such as abdominal discomfort, early satiety, and pain often arise, reflecting the physical consequences of an enlarged spleen. Furthermore, the heightened sequestration activity of the spleen exacerbates cytopenia, intensifying anemia and other blood cell deficiencies [42]. In therapeutic considerations, splenectomy emerges as a potential intervention to alleviate symptoms and ameliorate blood counts in select cases [43]. However, this approach is not without risks and is generally reserved for specific situations where the benefits outweigh potential complications. The labyrinthine role of the spleen in MF necessitates a deeper understanding of its pathophysiological transformations for the formulation of targeted and effective therapeutic strategies.
Fig. 2
Representation of the spleen’s role as a tumor surrogate in MF due to the engraftment of clonal cells. This diagram emphasizes the spleen’s compensatory function in hematopoiesis, leading to splenomegaly, EMH, and potential leukemic transformation, underscoring the spleen’s involvement in disease progression
Therapeutic approachesTherapeutic interventions for MF are designed to address the fundamental pathogenic mechanisms, alleviate symptomatic burden, and enhance the overall well-being of afflicted individuals. Among the pivotal strategies employed, JAK inhibitors, with Ruxolitinib at the forefront, assume a central role by targeting the deregulated JAK-STAT signaling pathway [44]. By mitigating constitutional symptoms, reducing splenomegaly, and enhancing quality of life, these inhibitors represent a significant advancement in disease management, particularly for those classified as intermediate- and high-risk. Beyond JAK inhibition, supportive care measures encompass interventions such as blood transfusions, erythropoiesis-stimulating agents, androgens, immunomodulatory agents (Thalidomide, Lenalidomide), and Prednisone which prove essential in managing anemia [45], whereas the use of Hydroxyurea (Carbamide) to manage leukocytosis and thrombocytosis, contribute to holistic disease management [46]. Allo-HSCT stands as the singular curative option, albeit constrained by factors like age and comorbidities, primarily reserved for younger patients with high-risk profiles [47]. Notably, newer JAK inhibitors, such as Fedratinib and Pacritinib, provide alternative therapeutic avenues, showcasing efficacy in specific patient subsets [48, 49]. A recently approved JAK inhibitor is Momelotinib which is an ATP-competitive small molecule inhibitor, that exerts its action by selectively targeting JAK1, JAK2 or JAK3, and TYK2 protein kinases, while also modulating activin A receptor type 1 (ACVR1), also known as activin receptor-like kinase 2 (ALK2), which represent pivotal clinical targets [50]. In Europe, the authorization of Momelotinib was granted on January 29, 2024, based on data derived from the MOMENTUM (NCT04173494) pivotal phase III trial [51], alongside a cohort of adult patients displaying moderate to severe anemia (hemoglobin < 10 g/dL) extracted from the SIMPLIFY-1 (NCT01969838) phase III trial [52]. Notably, Momelotinib secured approval from the FDA on September 15, 2023 [53], predicated on data emanating from same clinical investigations. This regulatory green light was for the treatment of anaemic patients afflicted with high/intermediate risk MF, encompassing both primary and secondary MF variants [54].
The exploration of combination therapies involving Ruxolitinib alongside histone deacetylase inhibitors or immunomodulatory drugs underscores ongoing endeavors to optimize treatment outcomes [55]. Combination therapies for MF are being actively investigated to enhance treatment efficacy by targeting multiple pathways involved in the disease. JAK inhibitors, such as Ruxolitinib, remain a key component of therapy, often used in combination with other novel agents to improve patient outcomes. For example, combining Ruxolitinib with Selinexor, an XPO1 inhibitor, has shown promise in reducing spleen size and alleviating symptoms by simultaneously targeting the JAK-STAT and nuclear export pathways [56]. Similarly, the combination of Ruxolitinib with Pelabresib (CPI-0610), a BET inhibitor, targets epigenetic regulation and has demonstrated improved responses in patients who are refractory to JAK inhibitors alone [57]. Other promising strategies include combining Ruxolitinib with Navitoclax, a BCL2 inhibitor, to enhance apoptosis and reduce spleen size, particularly in patients with resistance to Ruxolitinib monotherapy [58]. In addition, antifibrotic agents like PRM-151 are being tested with Ruxolitinib to target both inflammatory and fibrotic components of the disease, showing potential to reduce BM fibrosis and improve blood counts [59]. Furthermore, immune modulators such as Pomalidomide combined with Ruxolitinib have demonstrated efficacy in managing anemia and reducing transfusion needs in patients with severe cytopenias [60]. Lastly, hypomethylating agents like Azacitidine or Decitabine, when used with JAK inhibitors, have shown promise in improving BM function and potentially modifying disease progression [59]. These combination therapies represent a comprehensive approach to managing MF, targeting various aspects of the disease to provide better outcomes for patients.
As well, ongoing research, including clinical trials, probes into novel agents and targeted therapies tailored to specific mutations or anti-fibrotic mechanisms [61]. Symptomatic management, encompassing pain control and anticoagulation due to heightened thrombotic risk, complements the therapeutic paradigm. Rigorous disease monitoring, entailing periodic assessments of blood counts, molecular profiles, and imaging studies, remains integral for gauging disease progression and treatment response. In the evolving landscape of MF therapeutics, a variegated and personalized approach is imperative, considering diverse patient characteristics and the burgeoning insights into the intricacies of the disease.
The intricate interplay between the BM niche and MF pathophysiology underscores the mottled nature of therapeutic strategies, necessitating a comprehensive understanding of the molecular intricacies within the microenvironment for effective clinical interventions.
留言 (0)