
記住我
Clinical specimens used for sequencing were obtained from patients with GLM who underwent surgery or biopsy at the Renmin Hospital of Wuhan University. Detailed clinical information of the patients is shown in Supplementary Table 1. The tissue scrolls and sections used for H&E staining and immunofluorescence staining were from formalin-fixed paraffin-embedded mammary biopsy specimens provided by the Department of Pathology, Renmin Hospital of Wuhan University. Paracancerous tissue (normal) was defined as histologically normal adjacent breast tissue. Patients with GLM, fibroadenoma, or breast cancer were diagnosed based on pathological evaluation of fine-needle aspiration biopsy or surgical specimens. None of the patients with fibroadenoma or GLM had a history of malignant tumors. Patients with breast cancer who received neoadjuvant therapy were also excluded. The complete clinical information for these patients is presented in Supplementary Table 4.
Informed consent was acquired from all patients at Renmin Hospital of Wuhan University. Because only retained bacteria from Zhongnan Hospital of Wuhan University were used in this study, informed consent was waived for these patients. The study protocols were approved by the Ethics Committee of Renmin Hospital of Wuhan University (WDRY2020-K194 and WDRY2021-K093) and the Ethics Committee of Zhongnan Hospital of Wuhan University (2022225 K). All medical records were collected by a team of professional clinicians. Clinicians collected clinical information from the electronic medical records of the hospital information system (HIS). After exporting patients’ existing medical records from the HIS, we developed a standardized data collection form by extracting key information, such as clinical testing and culture results. These data were independently reviewed by two researchers to ensure the accuracy of data collection. If key information could not be obtained from the electronic medical records, the researchers collected it through communications with the attending doctors and other medical staff. If the data were still missing, the field was recorded as “unknown.”
H&E stainingHuman and rat mammary specimens were fixed overnight in phosphate-buffered 10% formalin. After dehydration, the samples were incubated in paraffin at 58 °C overnight and cast into molds covered with paraffin. Tissue sections were then prepared at a thickness of 5–10 μm. Paraffin sections were rehydrated with a decreasing gradient of ethanol concentrations and then stained with hematoxylin solution (0.1% hematoxylin, 5% Kal[SO4]2, and 0.02% KIO3). Counterstaining was performed by incubating the slides in eosin solution (1% eosin). Slices were dehydrated and cleared in xylene before mounting with neutral gum. H&E-stained images were acquired using a Panoramic MIDI system (3DHISTECH, Budapest, Hungary).
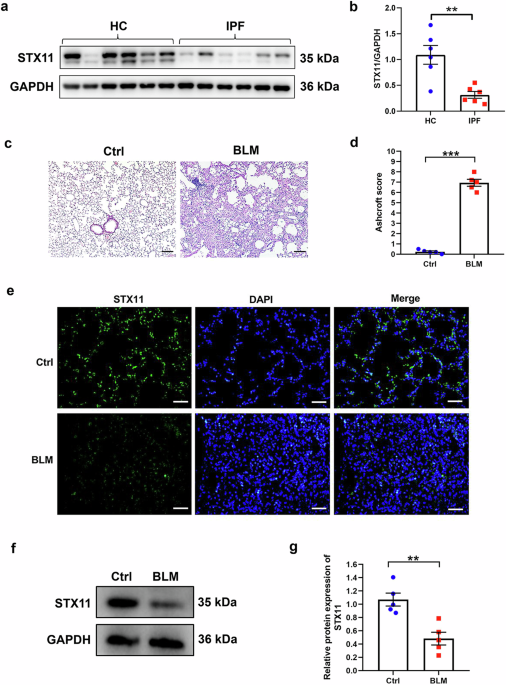
Immunofluorescence staining of tissues from patients and ratsNuclei were counterstained with 4′,6-diamidino-2-phenylindole. The images were captured using a panoramic MIDI slide scanner (3DHISTECH). Prior to immunofluorescence staining, mammary tissues extracted from animals and patients were fixed in 10% buffered formalin phosphate solution and embedded in paraffin according to standard protocols. Before antigen retrieval, the paraffin sections were dewaxed and rehydrated. Antigen retrieval was performed in ethylenediaminetetraacetic acid (EDTA) buffer (pH 8), and the sections were rinsed with PBS. The sections were then blocked using a blocking buffer for 30 min at room temperature and treated at 4 °C overnight in a humidified chamber with the primary antibody. Specific primary antibodies against IL-2 and IL-6 were obtained from Affinity Biosciences (OH, USA; AF5105, rabbit, 1:200; DF6087, rabbit, 1:200); IL-4 from Bo Ao Sen Biotechnology (Beijing, China; bs-0581R, rabbit, 1:100); IL-10 and IFN-γ from ProteinTech (Wuhan, China; 60269-1-Ig, mouse, 1:100; 15365-1-AP, rabbit, 1:100); and TNF-α from Boster Biological Technology (Wuhan, China; BA0131, rabbit, 1:100). After washing three times with PBS for 5 min each, the sections were incubated with a horseradish-peroxidase-conjugated secondary antibody for 50 min at room temperature in the dark. Quantitative image analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
16S rRNA metagenomic sequencingSamples were collected from patients and transported to the clinical laboratory within 2 h. All samples were then frozen immediately and stored at −80 °C prior to analysis. Each tissue sample was cut into small pieces using sterile scissors and homogenized. DNA was directly extracted from 200 μL of pus or pretreated rinse fluid using the QIAamp DNA Microbiome Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions.
The V3–V4 region of the bacterial 16S ribosomal RNA genes was amplified by PCR (95 °C for 3 min, followed by 35 cycles at 98 °C for 20 s, 58 °C for 15 s, and 72 °C for 20 s and a final extension at 72 °C for 5 min) using the barcoded primers, 341 F 5′-CCTACGGGRSGCAGCAG-3′ and 806 R 5′-GGACTACVVGGGTATCTAATC-3′. PCR was performed in a 30 μL mixture containing 15 μL of 2× KAPA Library Amplification ReadyMix, 1 μL of each primer (10 μM), 50 ng of template DNA, and ddH2O. Negative controls, consisting of empty sterile storage tubes, were processed for DNA extraction and amplification using the same procedures and reagents used for the tissue samples. No detectable amplification was observed in the negative controls. Amplicons were extracted from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s instructions and quantified using a Qubit® 2.0 instrument (Invitrogen, Carlsbad, CA, USA). All quantified amplicons were pooled to equalize their concentrations for sequencing using an Illumina MiSeq (Illumina, Inc., San Diego, CA, USA). Paired-end reads of 300 bp were overlapped on their 3′ ends for concatenation into original longer tags using PANDAseq (https://github.com/neufeld/pandaseq, version 2.9).
Bioinformatic analysisThe 16S rRNA sequences were clustered into operational taxonomic units using VSearch software and identity criteria of 97%. The Shannon index was used to compute the alpha diversity of the samples, and the Mann–Whitney U test was used to examine the differences in the Shannon index values among groups. We used the PERMANOVA test based on principal coordinate analysis with Bray–Curtis dissimilarity to compare the community composition of the microbiota among groups. The vegan package was used to perform alpha- and beta-diversity analyses in R. Linear discriminant analysis effect size was used to detect the taxa responsible for the different microbiota compositions. The logarithmic LDA cutoff score was set at 4.
Isolation and culture of C. parakroppenstedtiiAll the tissue or pus samples in our study were collected from patients with GLM who underwent routine culture in the clinic. The samples were incubated under aerobic and anaerobic conditions using one Columbia blood agar plate and one chocolate agar plate each at 35 °C. After observing the colony morphology of the microorganisms, suspected pathogenic microorganisms were selected for strain identification using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS analysis. For microorganisms that could not be differentiated by MALDI-TOF MS, biochemical reactions were used for strain identification. All 17 C. kroppenstedtii-like strains collected at the Zhongnan Hospital were isolated using routine culture methods.
For P1–P4 strain isolation, breast pus from the patients was used to inoculate BHIY medium (37 g/L brain-heart infusion broth and 10 g/L yeast extract) with 1% Tween 80, and 2% agar was added to make a solid plate. The plate was incubated at 37 °C in a 5% CO2 atmosphere for 48 h. The colonies on the plates were then isolated and identified as C. parakroppenstedtii using whole-genome sequencing. The bacteria were then preserved at −80 °C in 20% glycerol.
Subsequently, C. parakroppenstedtii was grown on BHIY with 1% Tween 80 culture plates to form single colonies. For subsequent interactions with animals or cells, one single colony was inoculated in 5 mL of BHIY with 1% Tween 80 liquid medium at 30 °C for 48 h, and all the bacteria were then transferred to 200 mL of BHIY with 1% Tween 80 liquid medium for culture at 30 °C until the optical density at 600 nm (OD600) = 2–3. Multiple vials of bacteria were cultured with the desired amount of cells and then collected by centrifugation and resuspended in PBS to a bacterial concentration of 1 × 105 colony-forming units (cfu)/µL, which was then used for subsequent animal experiments or cell experiments. C. parakroppenstedtii was inactivated by pasteurization for 40 min at 70 °C for obtain heat-killed bacteria, and follow-up culture was used to confirm that all bacteria were inactivated.
For subsequent corynekropbactin isolation experiments, several single colonies were inoculated in 10 mL of low-iron PGT medium43 or BHIY medium with 1% soy oil at 37 °C for 48 h, and then all the bacteria were transferred to 800 mL of low-iron PGT medium or BHIY medium with 1% soy oil for culture at 37 °C for 60 h. Multiple vials of culture supernatants were used for subsequent corynekropbactin extraction experiments. For corynekropbactin production analysis, several single colonies were inoculated in 20 mL of iron-free PGT medium with 1% soy oil at 37 °C for 48 h. Then, 1 mL of the bacteria were transferred to 100 mL of PGT medium with 1% soy oil and iron ions at different concentrations (Fe-free, 10 μM Fe2+, 10 μM Fe3+). After 80 h of fermentation at 37 °C, 50 mL of the culture broth was centrifuged at 7,000 rpm for 15 min for subsequent quantification.
To test the effects of different fatty acid sources on bacterial growth, single colony were delineated on PGT solid medium (low-iron PGT medium with 2% agar) with different fatty acid sources or on 14% parametrial breast cancer tissue.
Whole-genome sequencing of C. kroppenstedtii-like strainsThe genomic DNA of C. kroppenstedtii-like strains was extracted using a Bacterial Genomic DNA Extraction Kit (Tiangen, Beijing, China) following the manufacturer’s instructions. Whole-genome sequencing was performed and the genomes were assembled by GrandOmics (Wuhan, China) using the T7 (MGI, Shenzhen, China) and PromethION (Oxford Nanopore, Oxford, UK) platforms. To analyze genome similarity, the whole-genome sequences of the five strains were aligned in a pairwise fashion using bwa-mem to calculate similarities at the nucleotide level.
Cell experimentsThe human normal breast cell line MCF-10A was obtained from Procell (Wuhan, China) and cultured at 37 °C and 5% CO2 in MCF-10A cell special culture medium (Procell). The cells were maintained in the logarithmic growth phase. The medium was renewed every 2 d, and the cells were trypsinized with 0.25% trypsin-EDTA and subcultured in the same medium.
For interaction with C. parakroppenstedtii, MCF-10A cells were seeded in six-well plates (to measure cytokines) or T75 bottles (to detect corynekropbactins) and allowed to adhere overnight until the cell monolayer occupied ~90% of the vessel. In a six-well plate, 9 × 107C. parakroppenstedtii cells were added into each well (2 mL) and cultured at 37 °C and 5% CO2 for 24 h, or 1 × 106 live or heat-killed C. parakroppenstedtii cells were added to each well (2 mL) and cultured at 37 °C and 5% CO2 for 12 h. In a T75 bottle, 2 × 106C. parakroppenstedtii cells were added and cultured at 37 °C and 5% CO2 for 72 h. The cell culture supernatant was collected by centrifugation at 12,000 rpm for 10 min. One milliliter of supernatant from a six-well plate was collected and stored at −80 °C until further cytokine testing was performed, and 15 mL of supernatant from a T75 bottle was collected and stored at −80 °C until further corynekropbactins testing was performed.
For interactions with 1 and 2, cells were seeded in 96-well plates at a density of 4 × 103 cells/well. After 24 h, compounds 1 and 2 were added to the cells. Cytotoxicity was tested in MCF-10A cells using Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan). The blank control group was prepared by adding medium only or medium with 0.05% DMSO. Each group comprised six replicates. After 48 h, 80 μL of the cell supernatant was removed, and an equal amount of fresh medium was added to the same well. Ten microliters of CCK-8 reagent was then added and reacted for 2 h in the dark. The OD was measured at 450 nm using a microplate reader (BioTek, Winooski, VT, USA). Cell viability (%) was calculated as (OD treated - OD blank)/(OD control - OD blank) × 100%. The IC50 values for different drugs were determined using SPSS (IBM, Armonk, NY, USA). Subsequently, changes in cytokine levels were measured 6 h after the addition of 1 or 2 at their IC50 concentration (300 μmol/L and 50 μmol/L, respectively). Each group was comprised of four replicates. Eighty microliters of cell supernatants were removed and stored at −80 °C until further cytokine testing was performed.
Animal experimentsAll animal studies were reviewed and approved by the Laboratory Animal Welfare and Ethics Committee of Renmin Hospital of Wuhan University (Issue Nos. 20200702 and 20211001). Female Sprague–Dawley rats (8 weeks old for GLM-related experiments) used in this study were obtained from Hunan Silaike Jingda Laboratory Animal Co., Ltd. (Changsha, China). All animals were maintained on a 12-h light-dark cycle and housed in groups of three or four for at least 5 d before the experiments. All rats were provided with a standard laboratory diet and distilled water.
On the day of infection, the rats were anesthetized by continuous 2% isoflurane inhalation (1% oxygen). After anesthetization, approximately 1 × 107 cfu/rat of C. parakroppenstedtii or 100 μL/rat of PBS were injected as indicated orthotopically into the fourth mammary fat pad with an insulin syringe. The rats were sacrificed at the indicated time points (3, 8, and 10 d).
All rats were euthanized by carbon dioxide inhalation and blood was collected in a heparin solution by cardiac puncture, according to the experimental design. Mammary tissues were collected using sterile dissection tools, according to the experimental design. Infections were conducted in 5–12 animals per group, as indicated.
Analysis of cytokine levels in serum and cell cultureThe levels of the cytokines IL-2, IL-4, IL-6, IL-10, IFN-γ, and TNF-α in rat serum were measured using multiplexed sandwich enzyme-linked immunosorbent assays (ELISA) via custom-made magnetic Luminex multiple assays (LXSARM-06, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions, and the results were read on a Lumine X-200 instrument (Luminex, Austin, TX, USA).
The levels of TNF-α, IL-2, IL-4, IL-6, IL-10, and IFN-γ in MCF-10A cell culture were measured using an NMPA-approved kit (human Th1/Th2 subgroup detection kit; Cellger, Hangzhou, China) via a flow fluorescence assay (FACSCalibur, BD Biosciences, San Jose, USA) according to the manufacturer’s instructions.
Assay of iron-related factors in ratsRat serum samples were analyzed using an ADVIA 2400 Clinical Chemistry System (Siemens, USA) with Iron_2 reagents (Siemens Healthineers, USA). The total iron-binding capacity (TIBC) was measured by an ADVIA 2400 Clinical Chemistry System (Siemens) with TIBC reagents (Siemens). Transferrin saturation was calculated using the following equation: [total iron]/[TIBC] × 100.44 Ferritin levels were measured using an ELISA kit (Cusabio, Wuhan, China).45
High-performance liquid chromatography analysis of the bacterial supernatant and culture mediumThe culture supernatant and blank culture medium were absorbed by polystyrene-divinylbenzene copolymers (Amberlite XAD 16, Macklin) to enrich corynekropbactins and then eluted with methanol. High-performance liquid chromatography (HPLC) analysis was performed using an ODS column (ZORBAX SB-C18, 4.6 × 250 mm, 5 μm; 1 mL/min flow rate) with gradient elution. The UV spectra were recorded on a DAD instrument in the wavelength range of 200–600 nm. HPLC parameters were as follows: solvent A, H2O with 0.1% TFA; solvent B, 0.1% TFA in acetonitrile (ACN); gradient at a constant flow rate of 1 mL/min as follows: 0–60 min, 10–100% B and 60–70 min, 100% B; and detection by UV spectroscopy at 210 nm.
Extraction and purification of corynekropbactinsAfter fermentation of C. parakroppenstedtii P1 in PGT or BHIY medium with soy oil, the culture broth was centrifuged at 7,000 rpm for 15 min. The resulting supernatant was stirred with preprocessed XAD-16 resin under strain cultivation conditions for 2 d. The resin was then filtered and the material was eluted with pure methanol. The crude extract was concentrated under reduced pressure.
To purify corynekropbactins, the crude extracts were eluted using a normal-phase silica gel column. Fractions 18 to 24 (CH2Cl2:CH3OH, 0:1) were subjected to Sephadex LH-20 column chromatography and eluted with CH3OH to yield 40 fractions (10 mL/fraction). Fractions 8 to 15 were separated using semipreparative HPLC (ZORBAX SB-C18, 5 µm, 9.4 × 250 mm; gradient elution 0–60 min, 10–100% ACN and 60–70 min, 100% ACN; H2O with 0.1% formic acid [FA]; and flow rate, 3 mL/min) to yield compounds 1–8.
CAS assayCAS agar was prepared according to previously reported methods 46with minor modifications. To obtain 250 mL of CAS, the following reagents were used. Reagent A was prepared by mixing solution 1 (0.06 g of CAS dissolved in 50 mL of ddH2O) with 9 mL of solution 2 (0.0027 g of FeCl3 ∙ 6H2O dissolved in 10 mL of 10 mM HCl), which was then mixed with solution 3 (0.073 g of hexadecyl trimethyl ammonium bromide dissolved in 40 mL ddH2O) to obtain a blue dye, which was then autoclaved. Reagent B was prepared by dissolving 8.06 g of 1,4-piperazinediethanesulfonic acid in 225 mL of ddH2O. After adjusting the pH to 6.8, 3.75 g of agar was added, and the mixture was autoclaved and cooled to 50 ~ 60 °C. Subsequently, 25 mL of reagent A was slowly added to 225 mL of reagent B and mixed thoroughly. The solidified CAS agar plates were punched to a diameter of 5 mm, filled with 50 µL of sample, and then incubated at 37 °C for 2 d.
The ability of the corynekropbactins to chelate iron ions was determined using a previously reported protocol.47 For qualitative experiments, a liquid-type CAS assay was used. The CAS assay solution was prepared using the same method as that for CAS agar, but without the addition of agar. Sample solutions (100 µL) were mixed with the same volume of CAS assay solution in 96-well microtiter plates. To examine siderophores that induce a color change in the CAS solution, after 12 h of incubation at 30 °C, the OD630 nm values of the CAS solution with the solvent methanol (Ar) and CAS solution with compound sample (As) were measured using a Tecan Spark Spectrophotometer (Tecan, Groedig, Austria). In addition, the OD630 nm values of a series of two-fold dilutions of EDTA in methanol (12 µM–200 µM) were measured to draw a calibration curve to further compare the Fe(III)-binding activities with the compound samples.
Determination of the ability of corynekropbactins to chelate metal ionsFor metal chelating experiments, 60 µL of 600 µM purified corynekropbactin I, 400 μM corynekropbactin II, or methanol were incubated with an equal volume of 400 µM metal ion solutions (FeCl3, FeSO4, CuSO4, MnCl2, NiCl2, CoCl2) at 30 °C for 2 h, with 50% methanol used as the blank background. UV–vis spectra were recorded using a Multiskan GO microplate reader (Thermo Fisher Scientific) with a 250–1,000 nm range and 2 nm bandwidth.
Exploring the effects of ferric corynekropbactins on the growth of C. parakroppenstedtiiFor growth experiments, several colonies of C. parakroppenstedtii P1were inoculated in 5 mL of iron-free PGT medium with 1% Tween 80 liquid medium at 37 °C until OD600 = 2–3. The cultures were centrifuged at 3,000 × g for 15 min at 4 °C, and an equal amount of PBS was added to resuspend the bacteria, the sample was then centrifuged to remove the supernatant. After washing three times with PBS, an appropriate volume of PBS was added to resuspend the bacteria to OD600 = 0.5, and diluted 1:100 into each well of a flat-bottom 96-well plate containing 200 μL of PGT medium with 10 µM of iron, corynekropbactins, or ferric corynekropbactins. Ferric corynekropbactins were prepared by reacting 3 mM 1 or 8 with equal volumes of 3 mM FeCl3 or FeSO4 solution for 3 h. Excess iron ions within the reaction system were removed by semipreparative HPLC (ZORBAX SB-C18, 5 µm, 9.4 × 250 mm; gradient elution 0–60 min, 10–100% ACN and 60–70 min, 100% ACN; H2O with 0.1% formic acid [FA]; and flow rate, 3 mL/min). The corynekropbactins and their iron chelates were lyophilized, decontaminated with UV for 2 h, and diluted with iron-free PGT medium containing 1% Tween 80 to 10 µM. The plates were then incubated at 37 °C for 48 h in a Tecan Spark plate reader (Tecan Group Ltd. Switzerland), shaken at 220 rpm for 30 min every hour, and assayed for the OD600. Three replicates were used for each fraction, and iron-free PGT medium containing 1% Tween 80 was used as the control.
Antibacterial assaysFor agar diffusion assays, the appropriate agar medium (GM17 agar medium for L. lactis; MRS agar medium for Lactobacillus reuteri; LB agar medium for E. coli and C. glutamicum) was cooled to 50 ~ 60 °C and mixed with 1% overnight-cultured bacteria. The solidified bioassay plates (containing 25 mL of medium) were punched with a diameter of 5 mm with sterile punches and filled with 50 µL of sample. After incubation under the appropriate conditions (30 °C overnight for C. glutamicum, 30 °C for 2 d for L. lactis, 37 °C overnight for E. coli, or 37 °C for 2 d for Lactobacillus reuteri), the zone of inhibition in each well was observed.
The MIC was determined as previously described.48 Overnight-cultured bacterial strains were adjusted to 5.0 × 105 cfu/mL in culture medium (GM17 medium for L. lactis, MRS medium for Lactobacillus reuteri, and LB medium for C. glutamicum). A series of two-fold dilutions of each test compound was prepared. A total of 180 µL of the diluted bacteria and 20 µL of compound were mixed in 96-well microtiter plates and incubated under the corresponding conditions (30 °C overnight for C. glutamicum and L. lactis and 37 °C overnight for L. reuteri). Bacterial growth was monitored by measuring the OD600 nm using a Tecan Spark Spectrophotometer. The MIC was defined as the lowest concentration at which no bacterial growth occurred.
Liquid chromatography-tandem mass spectrometry analysis of the corynekropbactinsFifteen milliliters of cell culture were absorbed with polystyrene-divinylbenzene copolymers (Amberlite XAD 16, Macklin) to enrich corynekropbactins, and then eluted with methanol. The eluted methanol was concentrated to 2 mL for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
Breast tissue from rats was first cut into mung-bean-sized pieces, added to grinding beads, rapidly frozen in liquid nitrogen and quickly ground with a tissue grinder (65 Hz, interval of 10 s, repeated three times). Rapid freezing and grinding was repeated twice, and the tissue homogenate was obtained. Next, 1 mL of methanol was added for grinding and extraction, and the mixture was centrifuged at 10,000 r/min for 5 min to obtain the supernatant. All supernatant samples from same group were combined and concentrated to 200 μL for LC-MS/MS analysis.
Fifty milliliters of the bacterial culture broth supernatants were stirred with preprocessed 2% XAD-16 resin (w/v) under strain cultivation conditions for 8–12 h. The resins were filtered away and washed three times with deionized water, and the corynekropbactins were eluted with 6 mL of methanol and sonicated for 1 h. Two hundred microliters of eluent was used for for LC-MS/MS analysis.
LC-MS/MS datasets were acquired using a liquid chromatograph (Dionex Ultimate 3000, Thermo Fisher Scientific, CN) and a Q Exactive benchtop quadrupole-Orbitrap mass spectrometer (Q Exactive, Thermo Fisher Scientific), or a liquid chromatograph (ACQUITY UPLC H-Class PLUS, Waters, SG) and triple quadrupole mass spectrometer (TripleTOF 6600 + , AB Sciex, CA). Analytical-scale chromatographic separations were performed using a 2.1 mm × 150 mm reversed-phase column (Hypersil gold AQ, 2.1 × 150 mm column 0.2 mL/min) and binary solvent system comprising water (solvent A) and ACN (solvent B) gradient at a constant flow rate of 0.2 mL/min as follows: 0–30 min, 10–100% B; 30–50 min, 100% B. Negative-mode electrospray ionization was performed. The samples were analyzed in the m/z range of 50–1300 or 200–1500.
Antibiotic susceptibility of C. parakroppenstedtiiThe antibiotic susceptibility of the strains was determined using the Kirby–Bauer disk diffusion method, according to a previous study, with some modifications.49 McFarland turbidity was adjusted to 0.5, and the bacteria were inoculated using sterile cotton swabs on the surface of a Columbia blood agar plate (Dijing, Guangzhou, China). The following seven antibiotic disks (Oxoid, Hampshire, United Kingdom) were used: ciprofloxacin (5 µg), gentamicin (10 µg), metronidazole (5 µg), clindamycin (2 µg), rifampicin (5 µg), moxifloxacin (5 µg), and ceftazidime (30 µg). The antibiotic disks were placed on Columbia blood agar plates within 15 min of inoculation. The plates were incubated for 48–72 h at 30 °C in a 5% CO2 atmosphere. The diameters of the inhibition zones formed around the antibiotic disks were measured and interpreted according to the European Committee on Antimicrobial Susceptibility Testing guidelines (http://www.eucast.org).
Whole-genome phylogenetic tree methodMultiple sequence alignments (MSAs) generated using GTDB-Tk software (version 2.1.0)50 were used to construct a phylogenetic tree for the whole genome. IQ-Tree (version 2.2.0.3)51 was used to calculate the maximum likelihood phylogeny for the MSAs with the following parameters: -alrt 1,000, -bb 1,000, and -nt AUTO. The best-fitting model determined using ModelFinder,52 JTT + F + I, was well supported by the Akaike information criterion (AIC), corrected AIC (AICc), and Bayesian information criterion values. Tree branches were tested using an SH-like aLRT with 1,000 replicates. Finally, a phylogenetic tree was generated using the ITOL-v5 online tool.53
Exploring the influence of different washing agents on the growth of C. parakroppenstedtiiFifty milliliters of C. parakroppenstedtii broth (BHIY culture medium with Tween 80) in the logarithmic growth phase was centrifuged at 3000 × g for 15 min at 4 °C, an equal amount of PBS was added to resuspend the cells, and each sample was then centrifuged to remove supernatant. After washing three times with PBS, an appropriate volume of PBS was added to resuspend the pellet to an OD600 of 4. Subsequently, 100 µL of OD600 = 4 seed bacteria was transferred to 10 mL of BHIY culture medium with different washing agents (detergent, soap, or disinfectant) and cultured at 30 °C for 96 h. BHIY culture medium was used as the control group, and BHIY with Tween 80 culture medium was used as the positive control group. The cultures were then serially diluted 10 times, and 2 µL of each diluted culture was spotted on a plate with BHIY with 1% Tween 80 and cultured at 30 °C for observation for 48–72 h.
Exploring the influence of different drying methods on the growth of C. parakroppenstedtiiFifty milliliters of C. parakroppenstedtii broth (BHIY culture medium with Tween 80) in the logarithmic growth phase was centrifuged at 3000 × g for 15 min at 4 °C, an equal amount of PBS was added to resuspend the cells, and each sample was centrifuged to remove the supernatant. After washing three times with PBS, an appropriate volume of PBS was then added to resuspend the pellet to an OD600 of 4. Next, 5 mL of bacteria suspension was placed in a sterile Petri dish to determine the effect of different drying methods on bacterial activity.
A Petri dish containing 5 mL of C. parakroppenstedtii with an OD600 of 4 was placed in a biological safety cabinet. The lid was removed, and the plate was irradiated with a UV lamp (20 W, 30 cm) for 10 or 30 min. A non-UV lamp was used for the control group. The bacteria were then serially diluted 10 times, and 2 µL of each diluted culture was spotted on a plate BHIY with 1% Tween 80 and cultured at 30 °C for observation for 48–72 h.
A Petri dish containing 5 mL of C. parakroppenstedtii with an OD600 = 4 was placed in a 50 °C oven for 2 h to simulate common machine drying conditions. An appropriate volume of PBS was then added to supplement the evaporated liquid, and the volume in the Petri dish was restored to 5 mL. Next, the bacteria were serially diluted 10 times, and 2 µL of each diluted culture was spotted on a plate with BHIY with 1% Tween 80 and cultured at 30 °C for observation for 48–72 h.
Statistical analysisData are presented as the mean ± standard error of the mean, interquartile range, or standard deviation. Statistical significance was determined using the Kruskal–Wallis test, Wilcoxon signed rank test, Fisher’s exact test, and Dunn’s test, followed by the Benjamini–Hochberg correction, as appropriate. Two-sided P-values < 0.05 were considered statistically significant. Statistical analyses were performed using R (version 4.2.1), and graphical representations and data visualization were created using GraphPad Prism 8.2.1 (GraphPad, San Diego, CA, USA).
留言 (0)