
記住我
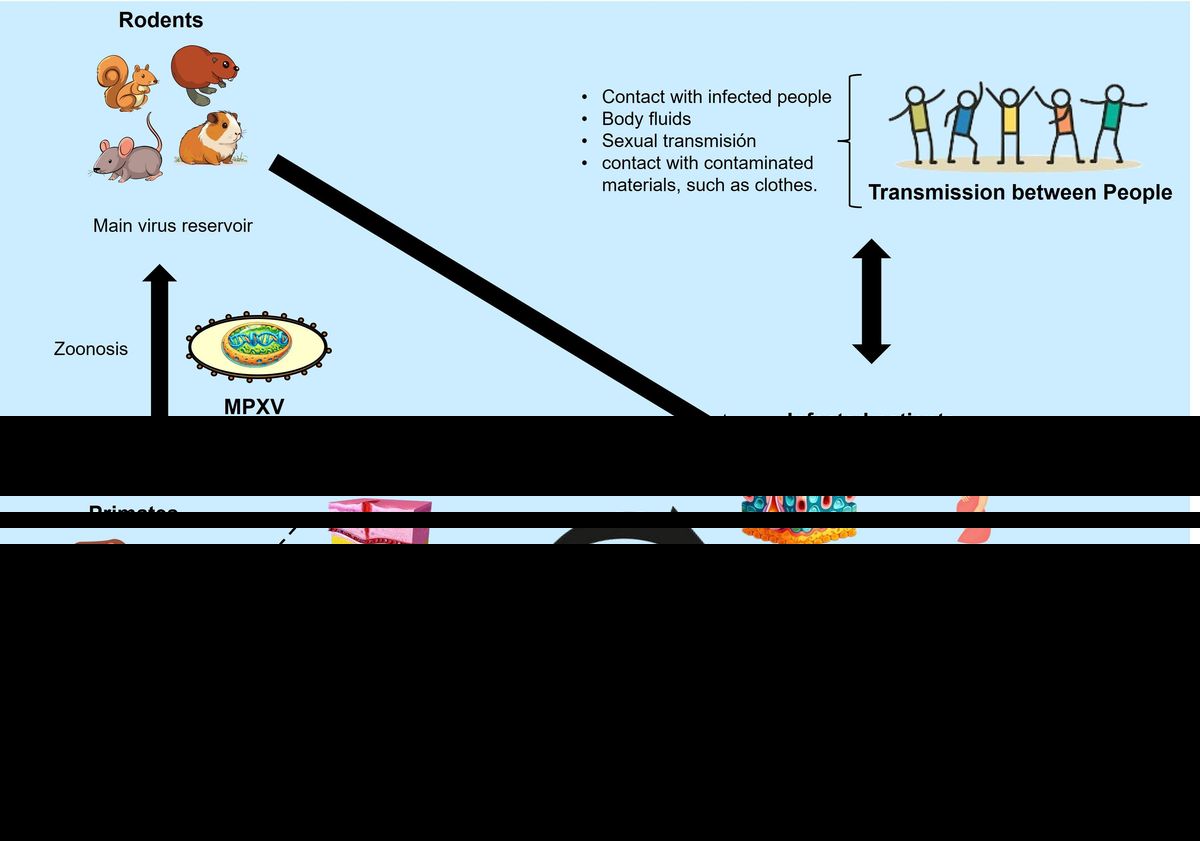





Monkeypox (mpox) is a zoonotic disease caused by the monkeypox virus (MPXV), which belongs to the Orthopoxvirus genus in the Poxviridae family (1). MPXV primary reservoirs are various species of African rodents, such as rope squirrels and rats of the genus Cricetomys. Through a zoonotic mechanism, the virus can be transmitted to primates via skin-to-skin contact, body fluids like blood or saliva, or sexual contact (Figure 1). The mpox disease manifests with symptoms similar to smallpox, though generally less severe, including fever, headache, muscle aches, and a distinctive rash that spreads across the body, reviewed in (2). The World Health Organization (WHO) has reclassified MPXV into Type I and Type II, formerly designated according to their endemic locations in Central and West Africa, respectively. This change in terminology aims to eliminate geographically discriminatory names (3). Type II MPXV is notably more virulent, with a fatality rate of 10.6%, compared to Type I, which has a fatality rate of 3.6%, as reviewed in (4).
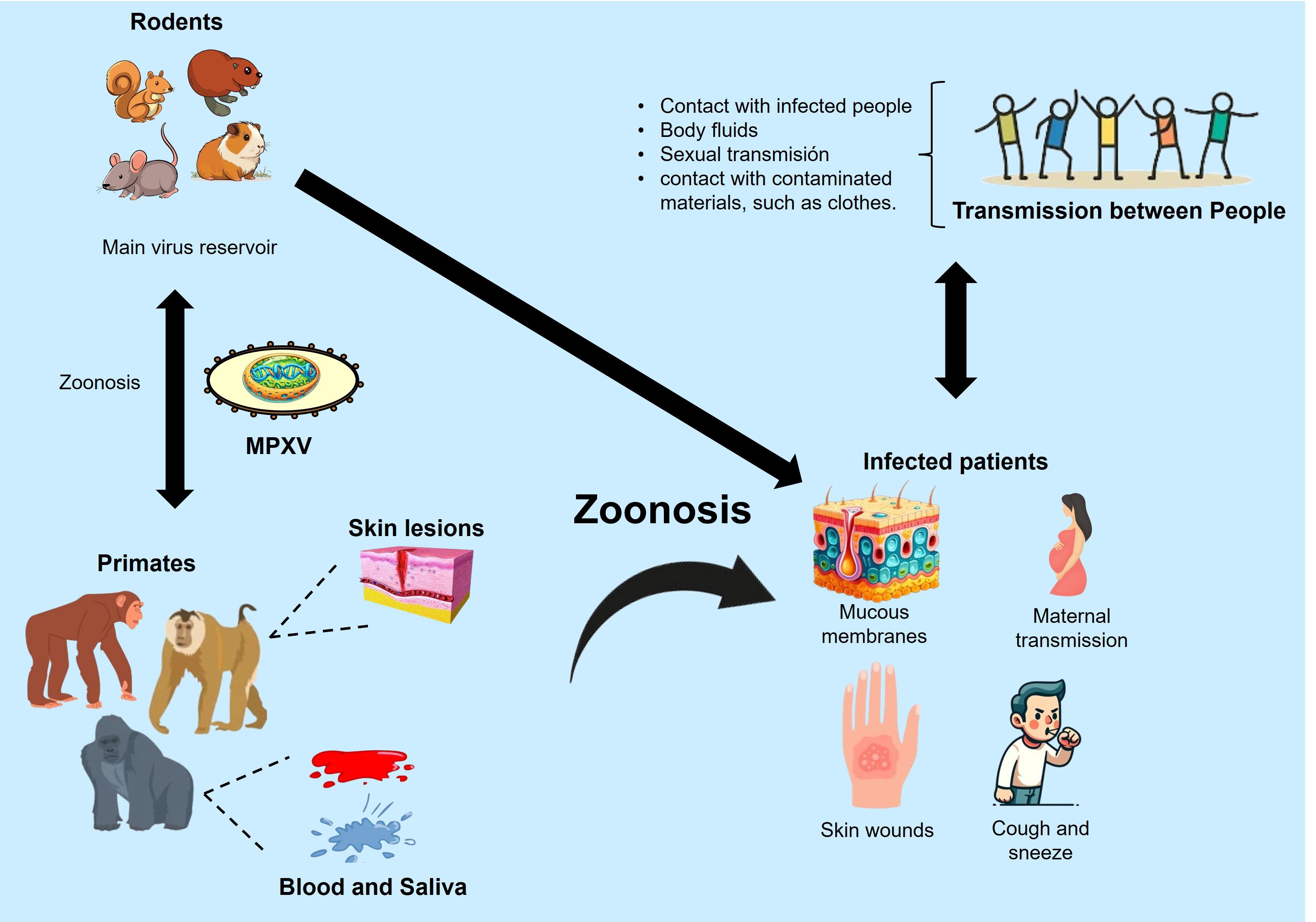
Figure 1. Zoonotic transmission mechanisms of MPXV. Modified from (106).
The recent emergence of outbreaks in 2024 has intensified global concerns about their potential spread. This outbreak and the one in 2022 affected non-endemic countries, underscoring the pandemic potential of emerging orthopoxviruses (5, 6). As of August 23, 2024, 3,331 confirmed human cases of mpox and 17,389 suspected cases had been reported on the African continent. The outbreak has resulted in 582 deaths, indicating an estimated fatality rate of 2.7% (7). The 2022 outbreak accelerated research and development of vaccines, but related publications significantly declined in 2023. However, by 2024, the issue has regained critical importance.
Recent genomic changes in MPXV, including insertions and deletions, have been reported (2, 8). These changes occur in conserved regions common across orthopoxviruses and sections specific to MPXV strains. These alterations affect the functionality of critical genes, such as those related to immune modulation and viral replication, contributing to the virus’s genetic diversity. Such adaptations could enable the virus to infect new hosts or thrive in different ecological conditions, increasing its pandemic potential (9). This underscores the importance of continuous surveillance and the need to be prepared by reinforcing research and developing new vaccination strategies to address future epidemiological threats.
Recently, groundbreaking vaccine candidates against MPXV have been developed, including cutting-edge multivalent mRNA and DNA vaccines based on identifying immunogenic proteins from the MPXV capsid. This review will delve into the cutting-edge advancements in mpox vaccine production, providing a critical overview of these emerging technologies and highlighting the urgent need to push the boundaries of research and development to create vaccines that can offer strong immune responses, durable protection, a significant reduction in symptoms, effective neutralization of the virus, and comprehensive disease prevention, all while ensuring safety for global populations.
2 Pathophysiology of mpoxMPXV enters the human body mainly through skin wounds, mucous membranes, or the respiratory tract. After entry, the virus replicates at the entry site before spreading to regional lymph nodes, where a second replication occurs. The virus then spreads from the lymph nodes to other organs via the bloodstream, leading to viremia, which distributes the virus throughout the body. Viral replication triggers an immune response that includes inflammation and the release of cytokines and chemokines, contributing to systemic symptoms such as fever, muscle aches, and general discomfort. A distinctive feature of mpox compared to other orthopoxvirus infections is lymphadenopathy (inflammation of the lymph nodes), which appears early in the disease. Complications and clinical symptoms of mpox are more common in children and immunocompromised individuals. They can include pneumonia, respiratory failure, sepsis, encephalitis, and corneal infection, leading to vision loss, reviewed in (10, 11).
Once the patient is exposed to the virus, the incubation phase begins, typically lasting between 7 and 14 days, with a maximum of 21 days. During this period, the virus replicates and spreads to the lymph nodes. Initial viremia leads to viral expansion and colonization of other organs. It is suggested that during this stage, the host enzyme APOBEC3, a cytidine deaminase capable of converting cytosine to uracil in exogenous DNA, may induce changes in the virus’s genetic material. These incidental changes could contribute to the virus’s genetic diversity and the emergence of new variants, increasing its pandemic potential, as reviewed in (12).
During the secondary viremia, symptoms begin and last between 0 and 5 days. This stage, known as the prodromal phase, is characterized by fever, headache, lethargy, myalgia, and lymphadenopathy, followed by a rash that lasts 2 to 4 weeks. The skin rash progresses through several stages: macules, papules, vesicles, pustules, and finally, crusts. These lesions are usually concentrated on the face and extremities but can appear anywhere. During this phase, serum antibodies become detectable, and patients can be infectious. The infection is typically treated with antiviral drugs such as cidofovir, ribavirin, tecovirimat, and brincidofovir, inhibiting viral DNA replication and development (13).
Most mpox cases are self-limiting, but complications can occur, especially in individuals with compromised immune systems. These complications can include secondary skin infections, pneumonia, sepsis, and even death. Infection resolution typically happens with the gradual recovery and shedding of scabs, often leaving scars. Immunity developed after infection may protect against future reinfections, although the extent and duration of this protection are not fully understood (14).
MPXV viral particles are oblong, measuring 200 to 250 nm in diameter. The virion consists of five distinct structures: core, membrane, lateral body, surface tubules, and nucleocapsid. The core has a bilobed shape and is surrounded by a double lipoprotein layer. Figure 2 shows the MPXV structure. The MPXV genome consists of double-stranded DNA comparable to smallpox. Its central region is 101 kb and shares 96.3% of the identity with the same Vaccinia Virus (VACV) area. The MPXV genome also has two variable terminal regions containing a 64 kb inverted terminal repeat (ITR) with some ORFs, hairpin loops, and short tandem repeats (15). It is 197 kb long and encodes at least 190 proteins, 30 of which are structural. Of the 190 proteins encoded by the MPXV genome, approximately 20 to 30 have been explored for potential use in vaccines. These proteins, particularly those located on the surface of the virion, such as A27L and B5R, have been studied due to their role in eliciting strong immune responses. These surface antigens are highly conserved across MPXV, VACV, and the smallpox virus, making them prime candidates for cross-protective vaccines. The conservation of these critical antigens across various orthopoxviruses enhances their potential utility in broad-spectrum vaccines, which could provide immunity against multiple poxviruses. Detailed studies have shown that these conserved antigens are crucial for the virus’s ability to infect host cells and are, therefore, integral to the design of effective vaccines (16).
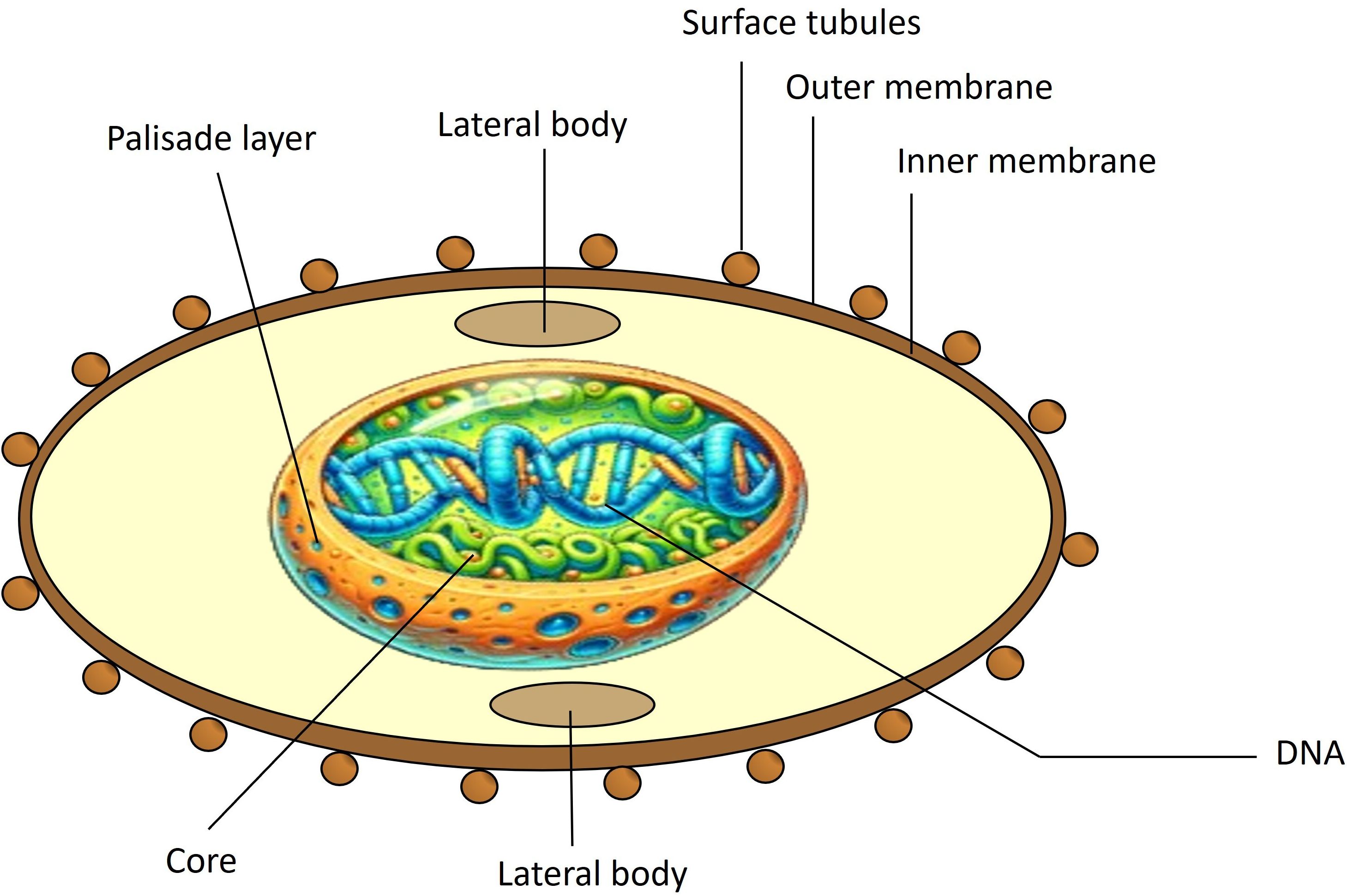
Figure 2. Structure of MPXV. MPXV consists of surface tubules, outer and inner envelopes of the extracellular virion, lateral bodies, a palisade layer (a protein layer in the core with parallel fibrils resembling a fence), and core fibrils.
Genomic differences between MPXV and VACV in genes coding for proteins that affect cytokines such as IL-1, tumor necrosis factor, and interferon can alter the host’s immune response, influencing the transmissibility and severity of infection. These genetic variations might explain why MPXV is not as fatal or transmissible as the smallpox virus but has the potential to become a more efficient human pathogen under certain circumstances, as reviewed in (17).
Replication, assembly, and maturation of MPXV occur in the cell’s cytoplasm, as shown in Figure 3. Virus replication involves three stages: early, intermediate, and late synthesis of mRNA and viral proteins, followed by virion assembly and morphogenesis. Virion assembly takes place in the host cytoplasm. MPXV has two infectious variants: the Intracellular Mature Virion (IMV), which is infectious when released from damaged cells, and the Extracellular Enveloped Virion (EEV), which buds from infected cells. These particles differ in membrane layers and surface glycoproteins, are active in infection, and can cause disease, as reviewed in (18).
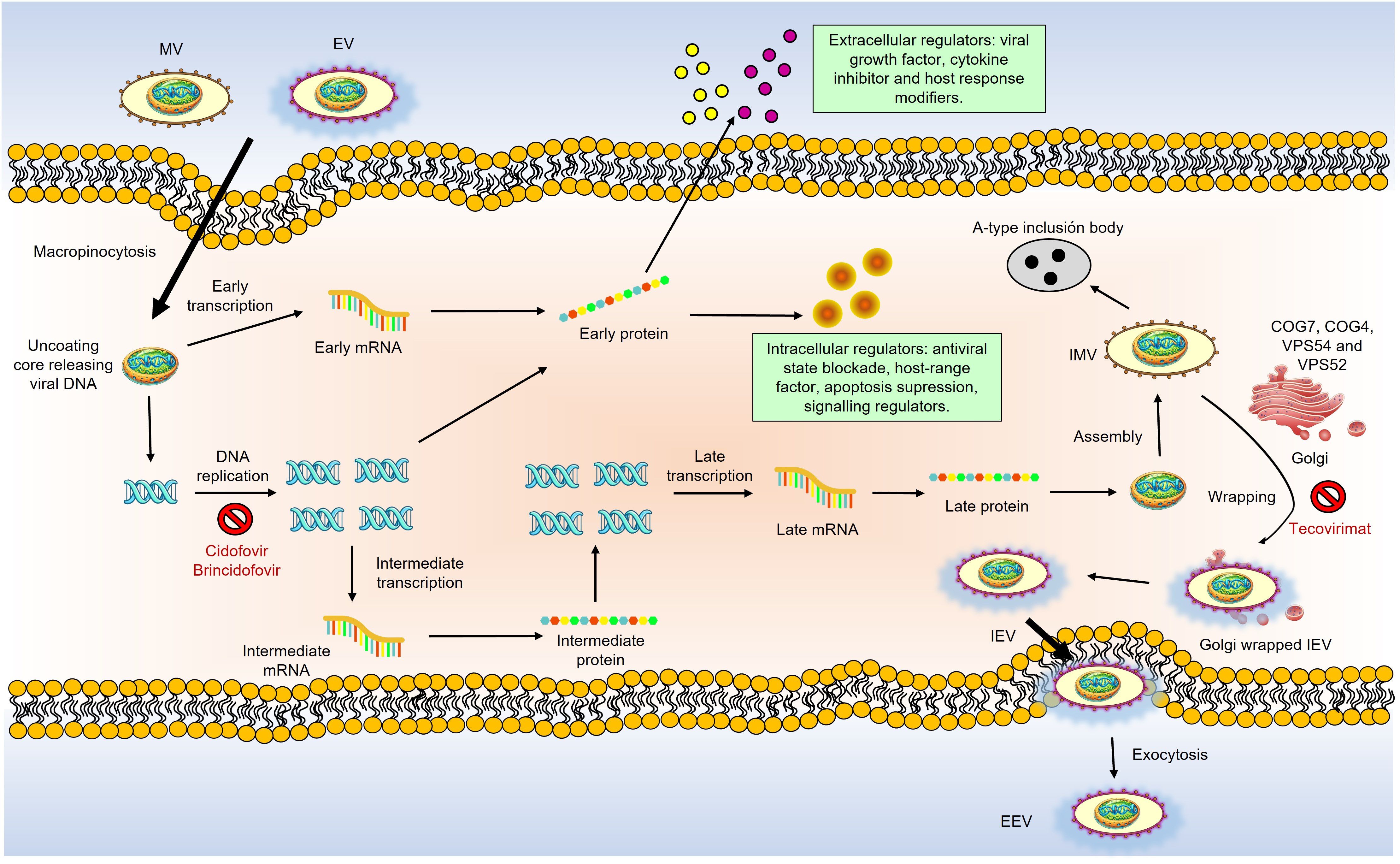
Figure 3. Schematic overview of MPXV life cycle and the action of anti-poxvirus drugs. MPXV replicates in the cytoplasm of infected cells. There are two infectious virions: intracellular mature virions (IMV) and extracellular enveloped virions (EEV), which differ in their membranes and glycoproteins. Virions bind to host cells via unknown receptors. MPXV replication involves early, intermediate, and late mRNA and protein synthesis stages, followed by virion assembly and morphogenesis. A double membrane from the Golgi wraps IMV to form intracellular enveloped viruses (IEV). GARP and COG complexes are crucial for infection, aiding in virus-cell interactions and the formation of EEVs. IEVs lose their outer membrane via actin-tail action, fuse with the cell membrane to become cell-associated enveloped viruses (CEVs) and are released as EEVs. MPXV also produces various modulatory proteins essential for replication. Anti-poxvirus drugs target different stages are pointed out in red: Cidofovir and brincidofovir inhibit viral DNA polymerase; tecovirimat and NIOCH-14 prevent CEV and EEV formation; Vaccinia Immune Globulin (VIG) prevents virions from infecting new cells.
Similar to VACV, IMVs are believed to be the most infectious form of the virus, entering cells via macropinocytosis and mediating host-to-host viral transmission (19). During infection, intracellular enveloped viruses (IEVs) form in the Golgi, shed their outer membrane layers via actin-tail action, and fuse with the cell membrane to produce cell-associated enveloped viruses (CEVs). CEVs are released to form EEVs, which enter cells through membrane fusion and facilitate viral spread within the host (20).
The key receptors for viral entry into host cells have not yet been identified, but conserved oligomeric Golgi complexes, COG7 and COG4, are known to be involved in viral entry and fusion. The Golgi-associated retrograde protein (GARP) complex also plays a significant role in virus-cell interactions necessary for viral entry and exit. In contrast, proteins VPS52 and VPS54 are responsible for forming extracellular enveloped virions and MPXV infection, as reviewed in (21, 22). MPXV infection activates T-cells and stimulates the release of inflammatory mediators through receptor interactions. However, this effect is not observed in individuals with a prior history of mpox, indicating the virus suppresses T-cell activation (23). These findings show that the mechanisms involved in MPXV infection are not yet fully understood.
3 Current and clinical-stage of vaccines for MPXVThe development of vaccines against MPXV primarily leverages the immune response generated by the VACV. This approach is grounded in two key factors: the significant cross-protection between VACV and MPXV and the conservation of crucial viral proteins. Antibodies induced by first-generation smallpox vaccines can effectively neutralize MPXV, offering long-term protection. Studies have always shown that over 97% of tested samples-maintained neutralization titers above the protective level of 1:32, indicating that most vaccinated individuals retain significant neutralizing antibodies decades after their last smallpox vaccination. This long-lasting immunity primarily targets conserved envelope proteins of extracellular VACV, such as A33 and B5, and mature virion proteins, such as L1 and A27, also present on MPXV. These proteins are crucial for virus neutralization and immune protection in animal and human models. Because these antigens are conserved across the orthopoxvirus genus, they are targeted in current vaccine strategies to elicit broad immune protection against multiple orthopoxviruses, including MPXV (24, 25).
Currently, three vaccines are approved for MPXV worldwide: JYNNEOS, LC16m18, and ACAM2000. JYNNEOS, approved in the USA and Mexico, is a live, non-replicating vaccine produced using a modified Vaccinia Ankara virus, incapable of replicating in human cells. This makes JYNNEOS a safer option, particularly for immunocompromised individuals. A case-control study in the United States estimated the vaccine’s effectiveness as 36% after a single dose and 66% after two doses (26). Another study reported that the vaccination series provided 85.9% effectiveness (27). JYNNEOS is generally well-tolerated, with common side effects including mild injection site reactions and transient systemic symptoms such as fever and fatigue (28).
Another approved vaccine, LC16m8, is used in Japan. It is a live attenuated vaccine derived from the Vaccinia virus. It was specifically developed to enhance safety by removing virulence-associated genes. Clinical studies have demonstrated the immunogenicity of LC16m8, showing a seroconversion rate of neutralizing antibodies against MPXV by day 28 post-vaccination (29). The vaccine has a favorable safety profile, primarily mild and transient side effects like injection site reactions and mild systemic symptoms like headaches and fever (30).
ACAM2000, approved in the USA, is a live, replicating vaccine derived from the Vaccinia virus. Although initially developed for smallpox, it is also effective against MPXV due to the cross-protective immune response elicited by the Vaccinia virus. Historical data on smallpox vaccinations, which demonstrated cross-immunity to MPXV, support the use of ACAM2000. However, this vaccine is associated with more significant side effects compared to non-replicating vaccines, including myocarditis, pericarditis, and generalized VACV infection. As a result, its use is limited to non-pregnant, immunocompetent individuals (31), and its risks are well-documented (32).
When evaluating the available vaccines for MPXV, it is essential to balance immunogenicity and safety. ACAM2000, JYNNEOS, and LC16m8 offer different profiles of efficacy and side effects, which are crucial in shaping vaccination strategies against monkeypox and other orthopoxviruses. ACAM2000, based on a live attenuated Vaccinia virus, provides robust cross-protection against smallpox and monkeypox. However, due to its replicating virus, it carries higher risks of side effects, particularly in immunocompromised individuals. JYNNEOS, utilizing a non-replicating Modified Vaccinia Ankara (MVA) virus, offers a safer alternative, especially for vulnerable populations, though it may induce a weaker immune response. LC16m8, derived from a highly attenuated Vaccinia virus strain, balances safety and immunogenicity, showing reduced side effects while eliciting a robust immune response. However, its availability is primarily limited to Japan. A comparison between these three vaccines is shown in Table 1.
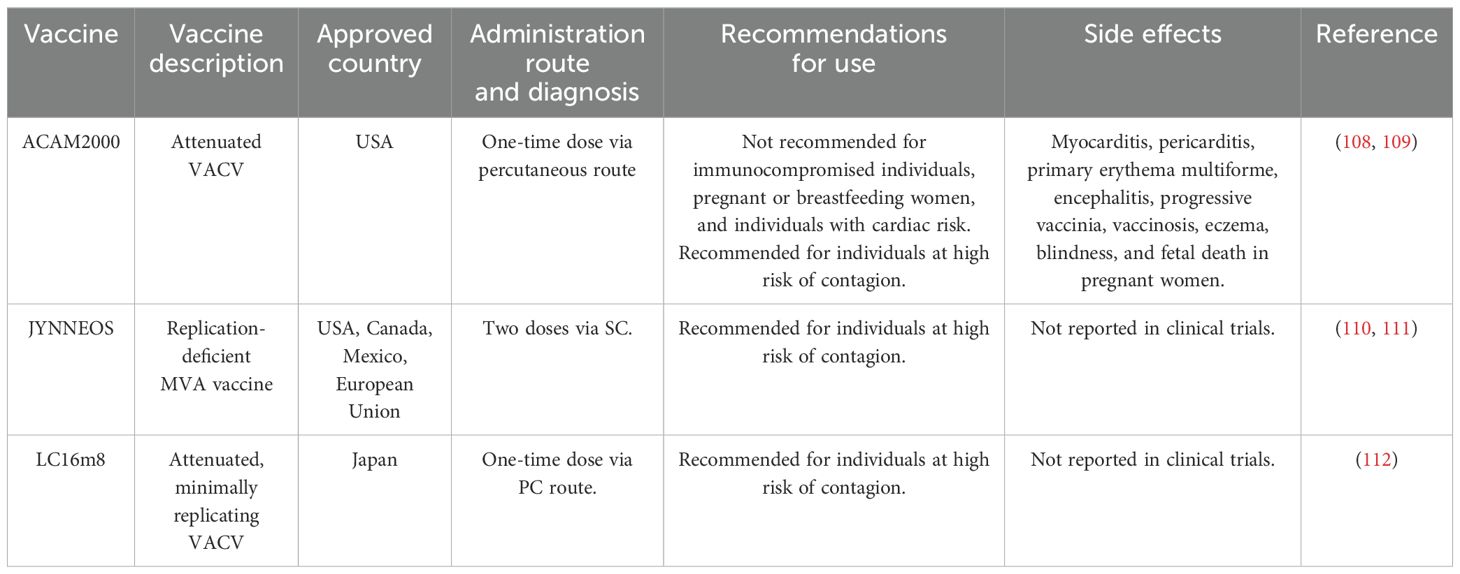
Table 1. Currently approved mpox vaccines.
A significant concern with attenuated virus vaccines like LC16m8 is the potential risk of reversion to a more virulent form. Although genetic modifications have been made to reduce this risk, there is no guarantee that the virus will not regain its pathogenicity under specific biological conditions (40). Additionally, attenuated viruses can recombine with endogenous viruses or other pathogens in the host, potentially creating new pathogenic viral forms that could evade immune control or existing treatments (33).
Non-replicating vaccines, such as JYNNEOS, also have their challenges, particularly in inducing a suboptimal immune response, especially regarding cellular immunity, which is crucial for protection against many viruses. This might necessitate multiple doses or the use of potent adjuvants to achieve the desired efficacy (34).
As of August 2024, the World Health Organization (WHO) recorded 22 clinical trials for MPXV vaccines, primarily based on attenuated or non-replicating viruses. These trials span Phases 1 through 4 and target diverse populations, including volunteers with no prior exposure to vaccinia, healthcare workers, men who have sex with men, and individuals living with HIV. Among the reviewed studies, only one (JPRN-jRCTs031220171) has reported results, while the remaining trials are either ongoing or recently completed. These studies aim to evaluate the safety, dosage, and efficacy of MPXV vaccines across various populations and contexts. It is worth highlighting a Phase I/II dose-escalation study is being conducted by BioNTech to assess the safety, tolerability, reactogenicity, and immunogenicity of the investigational RNA-based multivalent vaccine candidate BNT166a for active immunization against monkeypox (mpox) (NCT05988203). This trial consists of two substudies: Substudy A (SSA), involving approximately 48 vaccinia-naïve participants receiving two doses of BNT166a 31 days apart across three dose levels, and Substudy B (SSB), with 16 participants who have a prior history of smallpox vaccination (vaccinia-experienced), following the same dosing schedule. The trial began with a sentinel group in SSA, followed by dose escalation and expansion cohorts. While the initial design included a second candidate, BNT166c (trivalent), this arm was not activated. Table 2 summarizes the essential details of these clinical trials, including vaccine type, trial phase, vaccination scheme, target populations, and the availability of results.
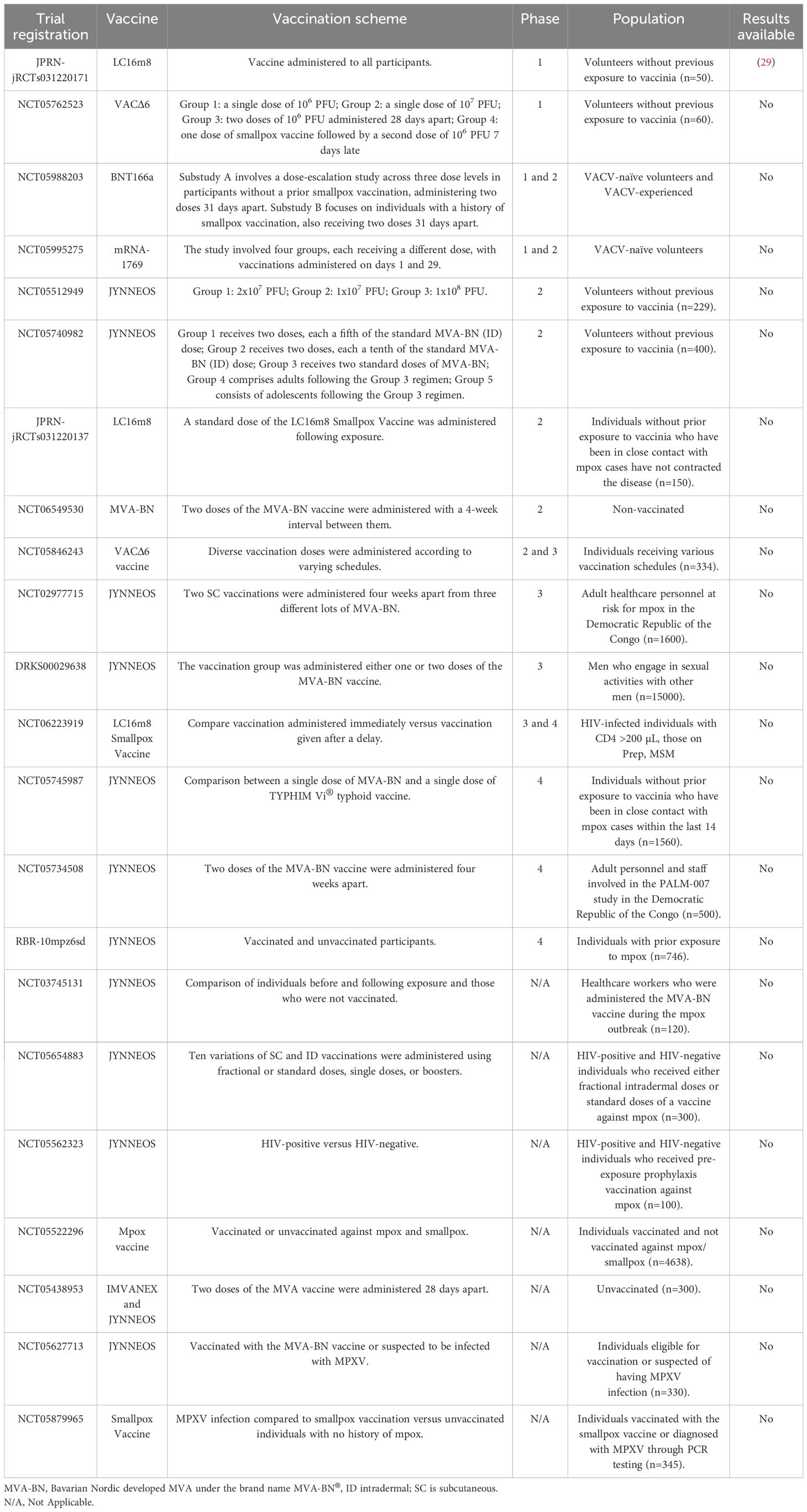
Table 2. Mpox vaccines in WHO-Registered clinical trials.
The analysis of vaccine platforms used in mpox development reveals distinct advantages and limitations across different technologies. Attenuated virus vaccines provide broad immune activation but carry risks such as reversion to virulence and require a cold chain. Non-replicating vaccines are safer for immunocompromised individuals but often elicit suboptimal immune responses. Viral vector-based vaccines offer durable immunity and can be developed rapidly, though they face challenges such as pre-existing immunity and complex manufacturing processes. Recombinant vaccines are safe and easy to store but may have lower immunogenicity without adjuvants. DNA vaccines are quickly developed and stable at room temperature but require advanced delivery systems. Lastly, mRNA vaccines are highly scalable and non-integrative but are associated with inflammatory responses and require an extreme cold chain for storage. Each platform must balance these trade-offs to optimize efficacy, safety, and logistical feasibility during vaccine development and deployment. Table 3 provides a comparative analysis of different vaccine platforms, highlighting their advantages and limitations.
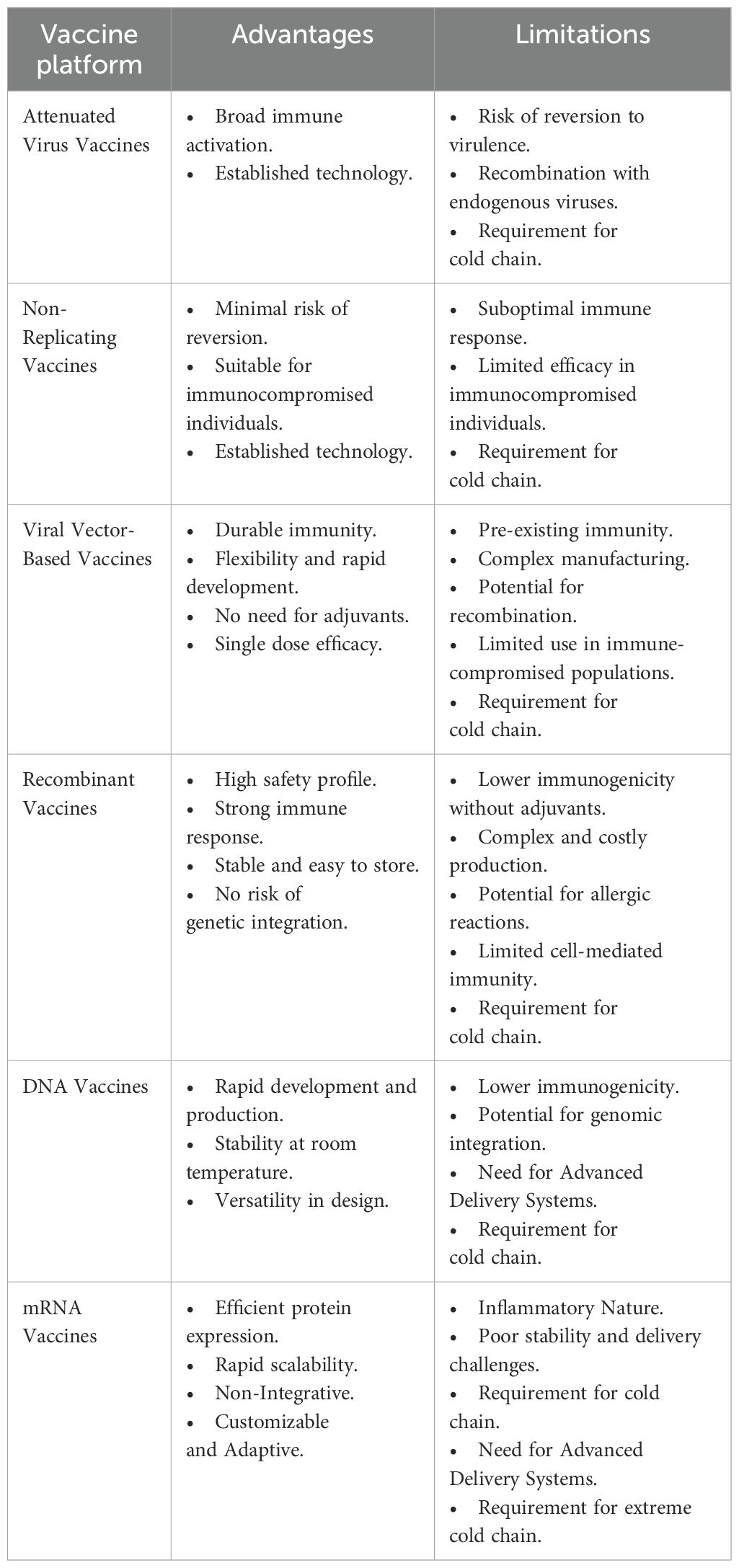
Table 3. Comparison of different platforms used in mpox vaccine development.
3.1 Vaccines based on attenuated or non-replicating virusesNew vaccine candidates against MPXV, based on attenuated or non-replicating viruses, are currently in preclinical stages. These vaccines introduce a safer form of the virus, which cannot cause disease but is sufficient to stimulate an immune response. Attenuated viruses are less virulent versions, while non-replicating viruses are designed not to replicate within the host. Table 4 discusses these preclinical vaccine candidates and reveals a variety of approaches, models, and outcomes.
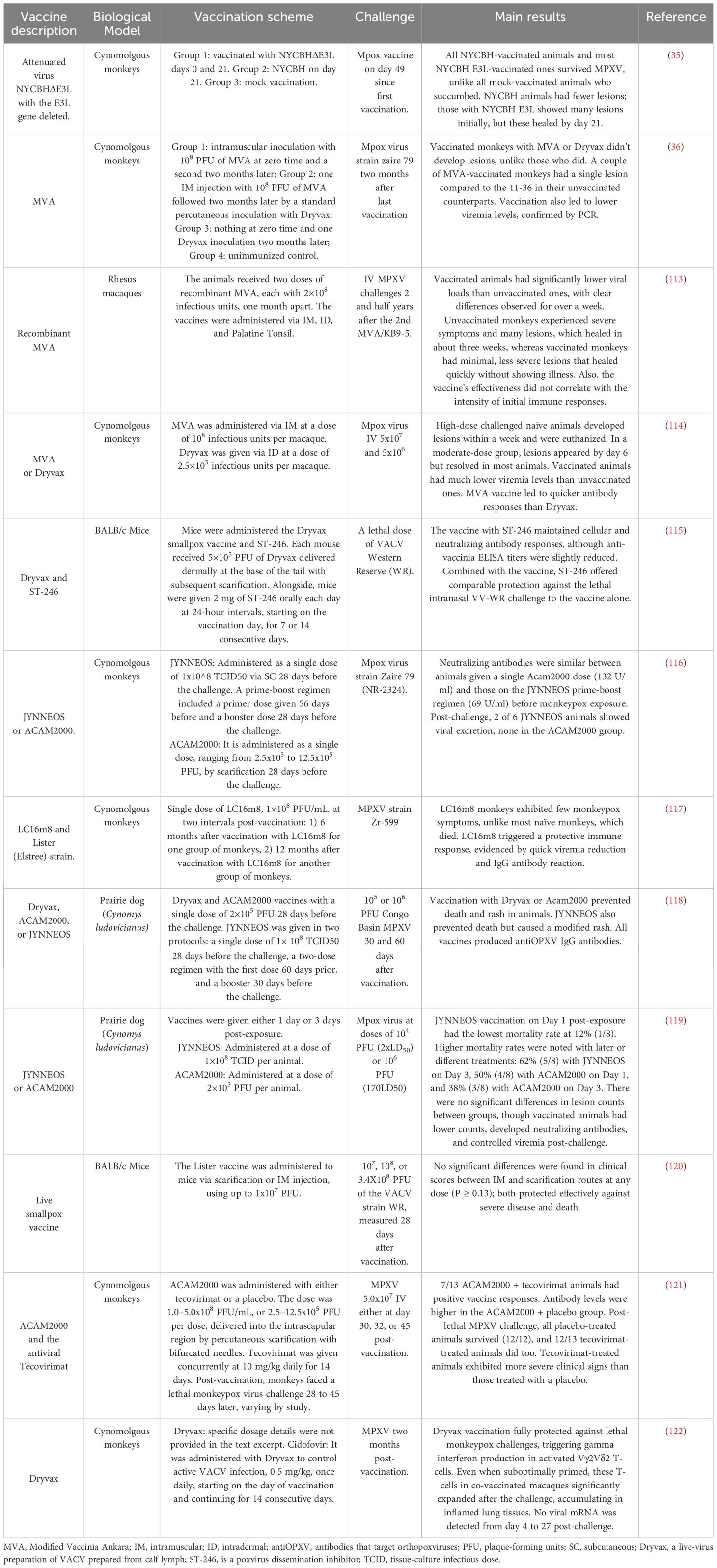
Table 4. List of preclinical mpox vaccine candidates based on attenuated and non-replicating viruses.
For instance, attenuated virus vaccines like NYCBHΔE3L, tested on cynomolgus monkeys, showed strong survival rates post-MPXV challenge, with fewer lesions than unvaccinated controls. Similarly, recombinant MVA vaccines showed efficacy in rhesus macaques by significantly lowering viral loads and resulting in minimal, quickly healing lesions (35).
In addition, Dryvax and ST-246, when administered to mice, maintained immune responses and protected lethal challenges, similar to the JYNNEOS and ACAM2000 vaccines tested in both cynomolgus monkeys and prairie dogs. These vaccines successfully induced neutralizing antibodies and controlled viremia, with some variations in post-exposure effectiveness depending on the timing of administration. Furthermore, live smallpox vaccines protected BALB/c mice against severe disease and death, regardless of the administration route. In cynomolgus monkeys, the combination of ACAM2000 with the antiviral Tecovirimat showed high survival rates after a lethal challenge, although tecovirimat-treated animals exhibited more severe clinical signs. Dryvax and Cidofovir also demonstrated full protection against MPXV in monkeys, highlighting a significant expansion of activated T-cells post-challenge, indicating a strong immune response (36).
The development of attenuated and non-replicating virus-based vaccines for mpox has made significant progress, notably with vaccines like ACAM2000 and JYNNEOS. ACAM2000 is effective but can cause serious side effects, including cardiac issues, as reviewed in (37). JYNNEOS, a non-replicating vaccine, has fewer complications, offering a safer and more effective option for mpox prevention (38). These vaccines, initially developed for smallpox, are identical in formulation but have been applied to mpox, demonstrating their effectiveness against both diseases and highlighting the adaptability of this approach for combating emerging viral threats.
Live virus-based vaccines have strengths and limitations. Their production is based on well-established technology, and they currently represent the majority of vaccines undergoing clinical evaluation. However, other types, such as mRNA vaccines, are also being tested, including BioNTech’s Phase 1 clinical trial (ClinicalTrials.Gov ID NCT05988203) (39). These vaccines can induce a robust immune response similar to a natural infection, effectively stimulating cellular and humoral immunity. However, drawbacks include the potential risk of reversion to virulent forms in attenuated viruses and the need for adjuvants or complex delivery systems for non-replicating viruses to be effective (40). Additionally, large-scale production of live virus vaccines is technically challenging and costly, requiring specialized biosafety facilities that comply with strict regulations to ensure safety, efficacy, and quality control. Facilities must adhere to Good Manufacturing Practices (GMP), which, besides being expensive, are crucial for controlling contamination and ensuring product consistency (41).
Given the risks associated with live virus vaccines, further research is essential to develop safer and more cost-effective vaccination platforms that facilitate universal access. The following sections will explore some of these emerging platforms in detail.
3.2 Viral vector-based vaccinesViral vector-based vaccines that do not use live viruses offer multiple benefits. They utilize non-replicating viruses, minimizing the risk of disease, which is particularly important for people with weakened immune systems. These vaccines can be designed to express specific antigens, generating a robust immune response without the risks associated with the use of live pathogens. This also allows for rapid adaptation to emerging pathogens. Additionally, they induce both humoral and cellular immune responses, protecting against viral infections (42, 43).
However, while viral vector-based vaccines can induce strong immune responses, the spectrum of the immune response may be limited. Depending on the vector and antigen used, some vectors may be more effective inducing a humoral reaction but less effective in generating a cellular response, or vice versa. One major challenge is preexisting immunity in the population. Many people may have preexisting immunity to the viral vector (such as adenoviruses), which can neutralize it before the viral antigen delivery, reducing its effectiveness (44). Additionally, the production and storage of these vaccines can be more complex due to the need to handle viruses.
The development of viral vector-based mpox vaccines has shown advantages inducing robust and long-lasting immune responses. Novel vaccine design technologies, such as reverse vaccinology and immunoinformatics, facilitate identifying and optimizing immunogenic epitopes derived from MPXV proteins. Table 5 shows some preclinical trials of viral vector-based vaccine candidates against mpox.
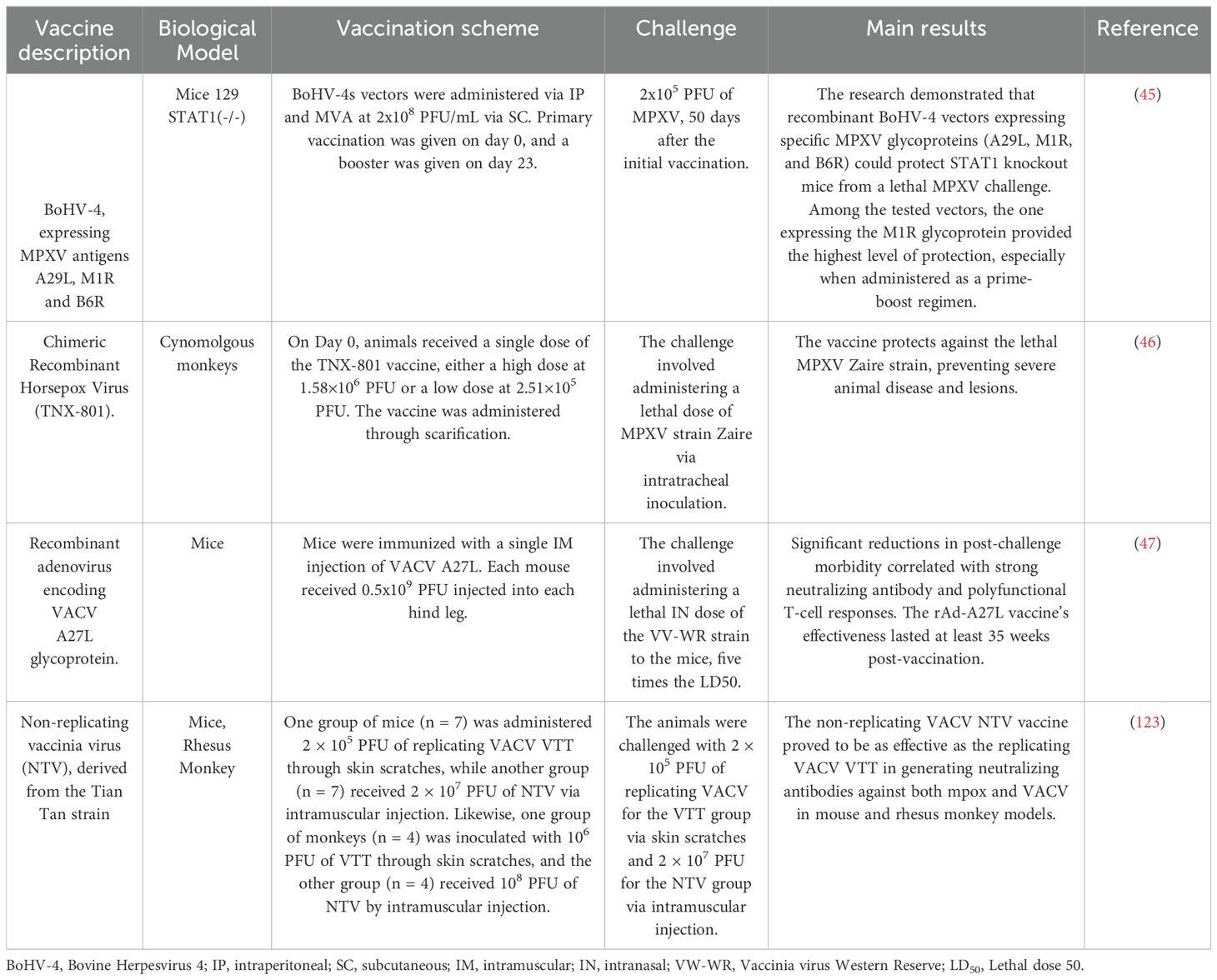
Table 5. Preclinical viral vector-based mpox vaccine candidates.
Comparing vaccine technologies across various studies reveals diverse approaches, biological models, vaccination schemes, and outcomes. One of the vaccines, the BoHV-4 Expressing MPXV Antigens vaccine, was tested in mice using intraperitoneal (IP) and subcutaneous (SC) vaccination routes. Results indicated that mice vaccinated with MVA were fully protected against the MPXV challenge, underscoring the critical role of the M1R antigen in achieving protection. However, the combination group (Combo/Combo) exhibited slightly lower protection rates, suggesting that a booster dose is essential for optimal efficacy (45). The Chimeric Recombinant Horsepox Virus (TNX-801), tested in cynomolgus monkeys, provided robust protection against a lethal MPXV Zaire strain, preventing severe disease and lesions (46). The Recombinant Adenovirus encoding VACV A27L Glycoprotein, tested in mice, significantly reduced morbidity after a lethal challenge, with sustained effectiveness for at least 35 weeks, indicating strong and long-lasting immunity (47).
Modified Vaccinia Ankara (MVA) vaccines, evaluated in multiple studies, consistently demonstrated reduced lesion development and lower viremia levels in vaccinated monkeys. MVA stands out as an effective platform for vaccine development. These vectors can deliver multiple selected epitopes that generate cellular and humoral immunity. Improving the immunogenicity of MVA-vectored vaccines has been a focal point in recent research, mainly through optimizing vector design and promoter utilization. One strategy involves deleting specific genes that modulate the innate immune response, which has been shown to enhance the quality and specificity of T-cell responses. For instance, the simultaneous deletion of the A44L, A46R, and C12L genes from the MVA genome resulted in significantly improved adaptive and memory T-cell responses, highlighting the potential for more effective vaccine vectors (48). Additionally, the use of stronger endogenous promoters, such as pF11 and pB8, has demonstrated a notable increase in transgene expression and overall immunogenicity, offering a promising avenue to enhance the efficacy of MVA-based vaccines, particularly in the context of emerging infectious diseases like mpox (49). These vaccines provide a robust safety profile and the ability to induce strong humoral and cellular immune responses, making them highly advantageous for use in diverse immunization strategies.
Building on these advancements, reverse vaccinology offers an innovative approach to refine vaccine design further. This method allows for incorporating T-cell and B-cell epitopes identified through immunoinformatics tools, enhancing vaccine specificity and effectiveness. By targeting critical immune components, reverse vaccinology complements the work done with MVA and other platforms, ensuring that vaccines elicit robust CD4+ and CD8+ T-cell responses, crucial for recognizing and eliminating infected cells. Additionally, potent B-cell responses lead to the production of neutralizing antibodies, providing long-term immunity. For instance, Bhattacharya et al. (2022) utilized immunoinformatics methods to design a vaccine targeting multiple virus epitopes, optimizing its ability to induce a solid and comprehensive immune response (50). Current vaccine strategies, including those for viral vector-based vaccines, have demonstrated both T-cell and B-cell mediated responses, contributing to their protective efficacy. While this reverse vaccinology approach has not yet been explicitly tested in viral vector-based vaccines, the principles of epitope design could be applied to enhance the effectiveness of such vaccines. Similarly, Ullah et al. (51) demonstrated how immunoinformatics analyses can predict vaccine constructs’ stability, immunogenicity, and non-allergenicity, which could also be leveraged to improve the formulation of viral vector-based vaccines (51).
Integrating these approaches accelerates vaccine development and creates safer and more effective vaccines for combating monkeypox and other emerging pathogens. Combining these innovative methods promises to transform the vaccine landscape as we progress, offering more adaptive and targeted strategies to combat viral outbreaks.
3.3 Recombinant protein-based vaccinesRecombinant protein-based vaccines utilize specific pieces of a pathogen, such as proteins, produced through genetic engineering techniques. These proteins are recognized by the immune system as foreign, triggering an immune response without the need for live or inactivated infectious agents, as reviewed in (52). Since they do not use the complete pathogen but only specific parts, they have a higher safety profile, with no risk of reactivation or causing disease. The most promising vaccine candidates are designed to induce strong immune responses by presenting viral antigens in a way that engages Toll-like receptors (TLRs) and the major histocompatibility complex (MHC), both of which are crucial for effective immune system activation. TLRs recognize pathogen-associated molecular patterns (PAMPs), including viral antigens or their molecular components. By incorporating adjuvants or specific viral components that mimic these PAMPs, the vaccine formulation ensures that the viral antigens are detected by TLRs, thereby triggering an innate immune response. This initial activation facilitates the presentation of antigens by MHC molecules to T cells, further amplifying the adaptive immune response. Additionally, they have good stability and do not require special storage conditions like live attenuated vaccines, as reviewed in (53).
The development of recombinant protein-based vaccines against mpox is progressing, with several studies evaluating their efficacy and safety. Current approaches include using specific viral proteins that induce protective immune responses. Preclinical studies on recombinant protein-based vaccines against monkeypox are shown in Table 6. Comparing vaccine candidates in various studies reveals diverse approaches, biological models, vaccination schemes, and outcomes.
Table 6. List of Preclinical Recombinant Protein-Based Mpox Vaccine Candidates.
The Subunit Smallpox Vaccine with VACV Proteins A33, B5, L1, A27, and CpG/Alum was tested in cynomolgus monkeys. In this study, Group 1 received three doses of 100 µg each at weeks 0, 4, and 12, while Group 2 received two doses at weeks 0 and 4. The challenge involved administering a lethal dose of MPXV five weeks after the final dose for the three-dose group and four weeks after the second dose for the two-dose group. The results showed that all animals in the three-dose group survived, while one animal in the two-dose group did not. The disease signs were least frequent in the three-dose group, and the vaccine was effective in enhancing neutralization titers and significantly reducing viral loads compared to controls (54).
Another study evaluated a Recombinant Subunit Smallpox Vaccine with A27V, A33V, B5V, and Adjuvants in BALB/c mice. The mice were vaccinated with 2 µg or 10 µg doses in histidine buffer and Alhydrogel, with an optional CpG adjuvant, and boosted two weeks later. Following vaccination, the mice were challenged intranasally with 1.7×105 PFU of VACV. The results indicated that all groups showed minimal weight loss and high survival rates (80-100%). However, the A27V formulation, despite eliciting strong antibody responses, did not effectively protect against infection (55).
The Polyvalent Vaccine with MPXV Proteins A29L, M1R, A35R, and B6R was also tested in BALB/c mice. In this study, mice were immunized with a recombinant protein vaccine combined with 15 mg of QS-21 adjuvant, following a three-dose regimen with an initial dose on day 0, followed by boosters on days 21 and 42. The challenge involved administering a lethal dose of the MPXV strain WIBP-MPXV-001 (2.24x108 PFU/mL) intranasally and intraperitoneally after the final immunization. The results showed that the vaccine significantly increased antibody levels, enhanced IFN-γ production, and improved Th1-mediated cellular immunity. Additionally, it effectively reduced MPXV replication and minimized organ damage in the mice (56).
The Escherichia coli-Expressed VACV A27L, B5R, and D8L vaccine was tested in BALB/c mice. In this study, mice were immunized with Escherichia coli-derived proteins using monophosphoryl lipid A and trehalose dicorynomycolate or TiterMax Gold adjuvants. The challenge involved intranasally administering 20 times the 50% lethal dose (LD50) of VV-WR (approximately 2.4x106 PFU). The findings revealed that three immunizations with A27L, D8L, and B5R, or just A27L and B5R, produced strong antibodies and provided full protection against a lethal dose of VACV. The recombinant proteins effectively neutralized the virus and offered protection against lethal challenges (57).
Although recombinant protein-based vaccines against mpox represent a promising advancement in vaccine technology, there are limitations. Recombinant protein vaccines may require adjuvants to enhance their immunogenicity and often need multiple doses to achieve long-lasting immunity. Additionally, the production of these vaccines can be complex and costly. Future research should optimize these vaccines to enhance their efficacy, reduce costs, and streamline production processes, ensuring they can be widely accessible and effective in preventing mpox.
3.4 DNA-based vaccinesDNA vaccines work by introducing plasmids containing genes encoding specific antigens of a pathogen under the control of a eukaryotic promoter. These plasmids are administered into tissues, where the cells transcribe and translate the plasmid genes into proteins. The immune system recognizes these proteins as foreign, triggering an immune response that includes cytotoxic T-cell activation and antibody production. A key benefit of DNA vaccines is their ability to induce both cellular and humoral immune responses, which is crucial for pathogens requiring T-cell-mediated immunity, such as HIV and other intracellular agents. Genetic engineering allows the inclusion of genetic adjuvants, like immunomodulatory CpG motifs, to enhance the vaccine’s immunogenicity. DNA vaccines’ rapid design and production make them particularly useful for responding to emerging diseases and pandemics (58–60).
DNA vaccines have several advantages over other types, such as simplicity and cost-effectiveness, over traditional vaccines and viral vectors. Their design is straightforward, and they possess high stability, making them ideal for regions with limited access to refrigeration (61, 62). They are well-tolerated and can be administered repeatedly for prolonged protection without significant side effects. DNA vaccines have shown efficacy in preclinical models against various diseases, including viruses, bacteria, and parasites (63, 64). However, they typically induce less potent immune responses than conventional vaccines, so methods to improve their delivery and effectiveness are being explored, as reviewed in (65). For instance, adding immunostimulatory sequences, such as CpG and dsRNA motifs, to the transcribed region of a DNA vaccine can significantly enhance the Th1 response. These motifs can influence the production of Th1 and Th17 cytokines, improving the overall immunogenicity of the vaccine. In animal models, these modifications increase protection against pathogens like Mycobacterium tuberculosis (66).
A common concern with DNA vaccines is the potential integration of the introduced DNA into the host genome, which could lead to unintended genetic consequences such as insertional mutagenesis, disrupting normal cellular functions, or triggering oncogenesis (67). Nevertheless, studies on plasmid DNA vaccines in mouse models showed a shallow integration frequency into the host genome, often below detectable levels (68, 69). These studies indicated that while the risk exists, it is relatively low.
Significant advances have been made in DNA vaccines for monkeypox, particularly in designing multivalent vaccines and using intradermal electroporation techniques to improve delivery and efficacy. Some preclinical studies of DNA vaccines against monkeypox are shown in Table 7.
Table 7. Preclinical DNA vaccine candidates for mpox.
The comparison of DNA-based vaccine candidates across different studies highlights varied approaches, biological models, vaccination schemes, and outcomes. In one study, rhesus macaques were immunized with plasmid DNA encoding monkeypox versions of VACV proteins (L1R, A27L, A33R, B5R), delivered via intradermal (ID) and intramuscular (IM) routes, followed by a protein boost with either alum or CpG-B ODN 2006. The MPXV challenge was administered intravenously (IV) at 5x107 PFU five weeks after the last protein boost. Results showed that animals receiving DNA and protein exhibited mild, quickly resolving disease, with high antibody levels and controlled viremia. In contrast, animals that received only DNA developed lesions, had low antibody titers, and did not survive, while those given only proteins experienced moderate to severe disease but survived (70). In another study, a multivalent DNA vaccine containing eight VW-WR genes (A4L, A27L, A33R, A56R, B5R, F9L, H3L, and L1R) was tested in cynomolgus monkeys. One group received ID injections, while another received IM injections, with vaccinations on days 0, 28, and 56. A lethal dose of the Zaire 79 strain of MPXV was administered intravenously. All vaccinated animals survived, exhibiting controlled viremia and developing neutralizing antibodies, particularly in the ID group. In contrast, most control animals succumbed to severe disease, with only one surviving but showing disease signs (71). A separate study involving a smallpox DNA vaccine in rhesus macaques employed multiple gene gun vaccinations at weeks 0, 3, and 6, followed by an MPXV Zaire-79 strain challenge administered intravenously. The vaccinated monkeys survived but continued to shed the virus, with the 4pox DNA vaccine group showing minimal signs of mpox and developing neutralizing antibodies. Control animals quickly succumbed to hemorrhagic mpox (72). In mice, a smallpox DNA vaccine was administered using a skin electroporation device with four 30 µg plasmid doses delivered via a microneedle array at weeks 0, 3, and 5. The challenge involved intranasal administration of the VACV strain IHD-J at 2×106 PFU. Vaccinated mice produced antibodies against four target immunogens and survived the lethal challenge without severe disease, while all control mice died within 6 to 8 days (73). Finally, a DNA-based 4pox vaccine targeting the L1, A27, B5, and A33 proteins was tested in rabbits. Rabbits were vaccinated by intramuscular electroporation with either a low dose (0.4 mg) or a high dose (4.0 mg) of the vaccine, administered thrice at one-month intervals. The challenge involved aerosolized Rabbitpox Virus at approximately 1,000 PFU. Unvaccinated rabbits developed severe disease and were euthanized by day 7, while all vaccinated rabbits, regardless of dose, survived, developed antibodies, and controlled viremia (74). These studies collectively demonstrate the potential efficacy of DNA-based vaccines in eliciting strong immune responses and providing protection against MPXV and related viruses across various animal models.
Additionally, the design of multi-epitope vaccines using reverse vaccinology and immunoinformatics has led to promising vaccine candidates by in-silico simulations. These studies suggest that the candidates can generate robust immune responses and provide effective global population coverage, pending further in vitro and in vivo studies (50). The most promising candidates are designed to induce strong immune responses by presenting viral antigens to Toll-like receptors (TLRs) and the major histocompatibility complex (MHC), both crucial for effective immune system activation (75, 76).
Current approaches in developing DNA vaccines for mpox also evaluate their immunogenicity through immunological simulations to predict human body responses. These approaches aim to improve vaccine efficacy while reducing development time and associated costs (76).
3.5 mRNA vaccinesThe new generation of messenger RNA (mRNA) vaccines offers significant advantages over previous technologies. Unlike DNA vaccines, mRNA vaccines provide superior safety since mRNA, as a temporary information carrier, does not interact with the genome, reviewed in (77). Additionally, their production process does not require handling the pathogen; instead, it uses gene sequences obtained through in vitro transcription from a complementary DNA (cDNA) template using a bacteriophage RNA polymerase (78). Moreover, mRNA vaccines can induce cellular and humoral immune responses (79, 80). These vaccines express proteins in situ, triggering balanced cellular and humoral responses (79, 80).
The RNA’s ability to induce controlled cellular and humoral immune responses begins when an antigen-presenting cell endocytoses the mRNA. Double-stranded RNA (dsRNA) acts as PAMP recognized by various innate immune system receptors, such as TLRs. TLR3 and TLR7/8 in endosomes recognize the RNA and trigger a type I interferon (IFN-I) response, which is crucial for an effective antiviral response. This pathway involves proteins like RIG-I, LGP2, NOD2, RIG-1, MDA5, and PKR (81). MDA5 and PKR play critical roles in inducing IFN-I. MDA5 recognizes intracellular dsRNA, activating IFN-β transcription depending on the cell type. At the same time, PKR can induce apoptosis and is essential for MDA5-mediated signal transduction in certain viral infections, such as the VACV virus, and for regulating the immune response (82). The protein produced by the mRNA is then released from the cell to activate B-cells. Simultaneously, proteins generated from the mRNA, newly synthesized and re-endocytosed, are processed into peptides by the proteasome. These peptides are presented on MHC-I or MHC-II molecules, activating CD4+ and CD8+ T-cells, which in turn drive the activation of adaptive immunity (83–85) (Figure 4).
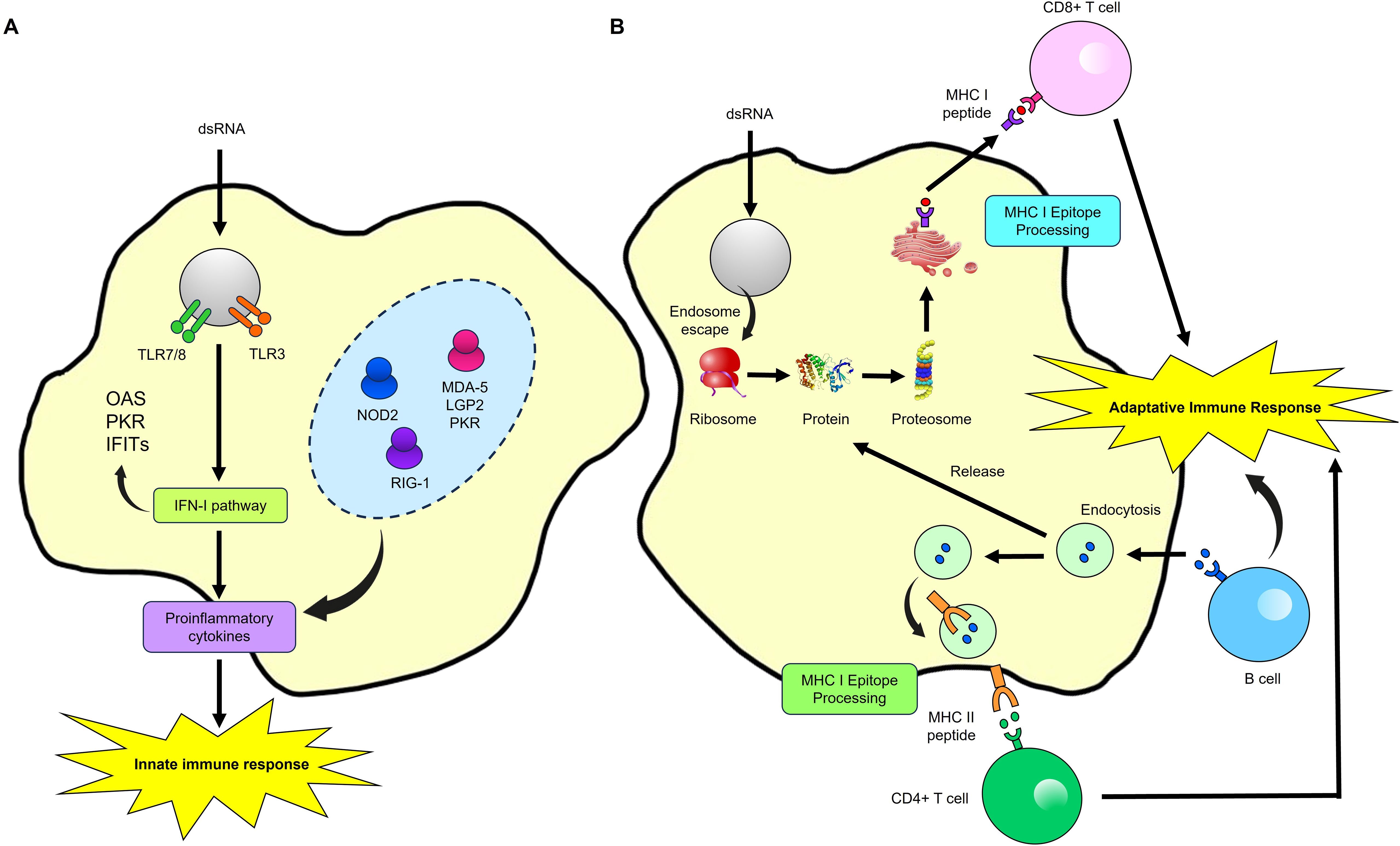
Figure 4. Immune Response activation by a mRNA Vaccine. (A) Innate immune response. When antigen-presenting cells endocytose exogenous mRNA, it is detected by TLR7/8 and TLR3 in endosomes and by LPG2, NOD2, RIG-1, NOD2, MDA-5, and PKR in the cytosol. This detection induces strong type I interferon (IFN-I) responses and the production of proinflammatory cytokines, thereby activating innate immunity. (B) Adaptive immune response. The protein encoded by the mRNA is released from the cell to activate B-cells. Concurrently, mRNA-encoded or re-endocytosed proteins are processed into peptides by the proteasome and presented on MHC-I molecules, activating CD4+ and CD8+ T-cells. Modified from (107).
RNA vaccines provoke more inflammation because, during mRNA translation, some mRNA molecules can fold back on themselves, forming double-stranded RNA (dsRNA), a byproduct recognized by the immune system as a PAMP. This dsRNA and other PAMPs activate strong immune responses by engaging innate immune receptors. The mRNA in these vaccines is designed to instruct cells to produce specific viral proteins, which are then recognized by the immune system, triggering an adaptive immune response. Unlike DNA vaccines, which generate a more balanced Th1/Th2 immune response, RNA vaccines typically induce a Th1-dominant response. While a Th1 response effectively protects against many pathogens, it may not be
留言 (0)