
記住我





Genomic instability, defined as an increased tendency of genomic alterations during subsequent cell divisions, is a hallmark of cancer and plays key roles in malignant transformation, cancer progression, and response to treatment (1, 2). The accumulation of mutations and genomic alterations can result in the activation of oncogenes, the inactivation of tumor suppressor genes, and the dysregulation of key cellular pathways, ultimately leading to transformation and uncontrolled cell proliferation. Through the acquisition of genetic diversity, genomic instability fuels tumor evolution and endows tumor cells with a survival advantage, enabling them to adapt to changing conditions and upon selective pressure (1, 2).
Nevertheless, genomic instability can have detrimental effects on cell viability. In addition to reducing tumor cell fitness through several cell-intrinsic mechanisms, genomic instability triggers a cascade of cell and non-cell autonomous inflammatory signaling pathways (3). This leads to the release of cytokines and chemokines to attract immune cells and the expression of immune-activating signals for the rapid elimination of DNA-damaged cells (4–6). Moreover, alterations in the DNA sequence or genomic rearrangements can lead to the generation of mutated proteins that will eventually be presented as neoantigens and activate T cells (7). Hence, immune surveillance is a critical barrier to tumorigenesis, especially in the context of genomic instability. As a result, cancer cells need to evolve and activate mechanisms to reduce immunogenicity and therefore avoid detection and elimination by the immune system.
Over the last few decades, our understanding of the mechanisms of immune evasion exploited by cancer has led to the development of effective immunotherapies, particularly immune checkpoint blockade (ICB) (8, 9). Even though pro-inflammatory signaling and genetic alterations have been correlated with response to ICB (8), durable response rates in genomically unstable tumors remain paradoxically low (10–14), limiting the use of those drugs and highlighting the need to develop novel complementary approaches. To do so, a thorough molecular understanding of the specific mechanisms of immune recognition and immune evasion of tumors with high genomic instability is crucial. This review focuses on the complex interplay between genomic instability and the immune system in the context of cancer. We summarize the main inflammatory consequences triggered by genomic instability, primarily through cGAS/STING activation, and how the different types of immune cells can recognize genomically unstable tumors. Finally, we discuss the main mechanisms of immune evasion exploited by tumors with defects in DNA repair and tumors displaying high levels of chromosomal instability (CIN) and resulting aneuploidy and their implications for cancer immunotherapy.
2 Cellular mechanisms of genomic instability in cancerGenome stability is tightly monitored by several mechanisms, which include the DNA damage checkpoints, the DNA repair machinery, and mitotic checkpoints. Defects at any of these steps can result in genomic instability. Thus, genomic instability can present itself in multiple different levels ranging from single nucleotide point mutations to complex structural and numerical chromosomal abnormalities.
2.1 Defects in DNA repairThe DNA damage response (DDR) is a complex network of highly conserved pathways that have evolved to sense and repair various forms of DNA damage (3). The DDR comprises a number of mechanisms to repair various forms of damage including base excision repair (BER), single-strand break repair (SSBR), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and nonhomologous end-joining (NHEJ). While deficiencies in any of those pathways can give rise to genomic damage and contribute to tumorigenesis (3, 15), here, we focus the role of MMR and HR on genomic instability.
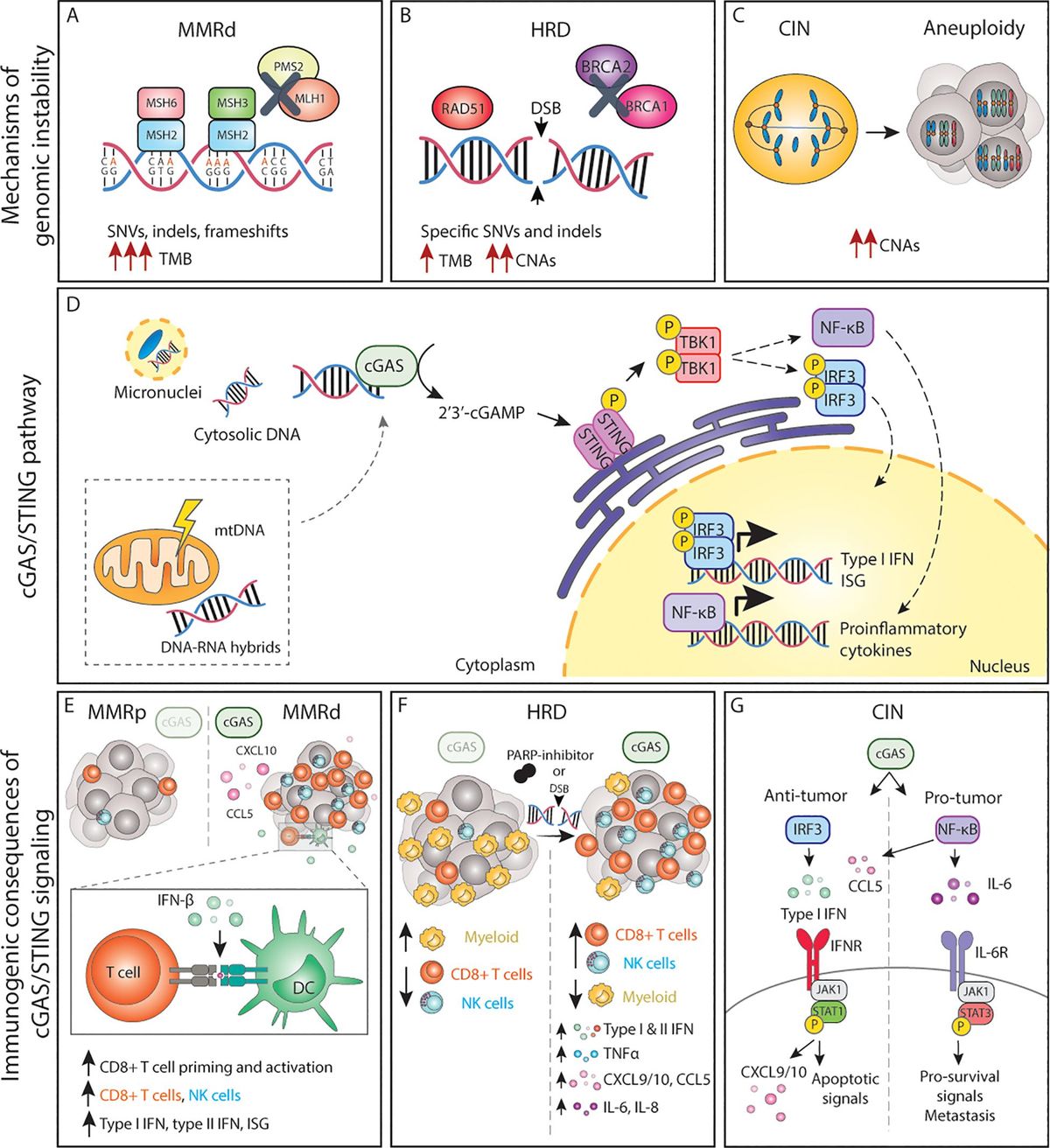
2.1.1 Defects in mismatch repairThe MMR pathway recognizes and repairs base pair mismatches and small insertions and deletions loops (indels) that occurred during DNA replication (16). Mismatched base pairs and small indels up to 3 nucleotides can be recognized by the MSH2/MSH6 heterodimer whereas larger indels are recognized by the MSH2/MSH3 complex (17, 18). Once these heterodimers recognize the error, a second complex formed by MLH1/PMS2 is recruited and forms a tetrameric complex. Subsequently, exonuclease 1 (EXO1) is recruited, activated, and removes the newly synthesized DNA strand, leaving a DNA excision gap and a region with a single-stranded DNA (ssDNA). DNA polymerases and ligases subsequently synthesize and seal the new correct DNA strand, repairing the damage (16). Although the mutation rate during DNA replication is low and DNA polymerases have proofreading activity and can correct some of these errors, others escape proofreading and require the MMR system for repair (16) (Figure 1A).
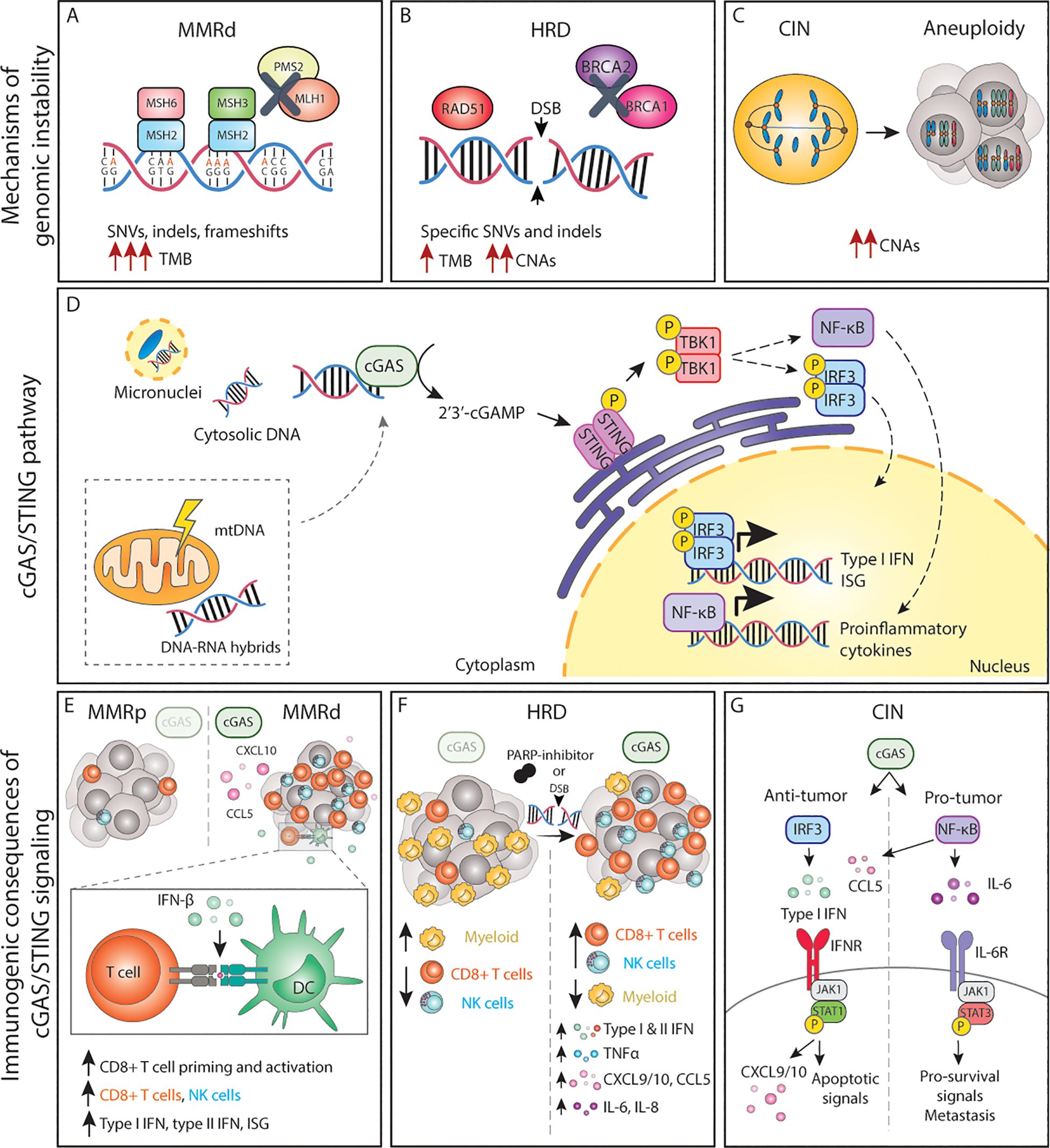
Figure 1. The cGAS/STING pathway and its inflammatory consequences in genomically unstable tumors. (A-C) Genomic instability can lead to the accumulation of dsDNA and other acid nucleic structures in the cytoplasm, which can activate cGAS. (D) Activation of cGAS either by nuclear DNA, micronuclei or other acid nucleic results in inflammatory signalling and expression of type I IFNs, ISG and NF-κB target genes. (E) In MMRd tumor, activation of cGAS results in increased CXCL10 and CCL5 production, which increase the number of tumor-infiltrating CD8+ T and NK cells as well as increased expression of IFN-β, which enhances DC-T cell interactions. (F) In HRD tumors, or tumors treated with DNA damaging agents such as PARP-inhibitors, activation of the cGAS pathways results in expression of the chemokines and cytokines, leading to a more inflamed TME with higher number of cytotoxic immune cells. (G) The cGAS/STING pathway in CIN/aneuploid cells has both pro and anti-tumor effects. The activation of the IRF3-type I IFN signalling axis results in immune surveillance and apoptotic signals, whereas NF-κB signalling mainly promotes IL-6/STAT3 pro-survival signals and enhance metastatic potential. MMRd, mismatch-repair deficient; MMRp, mismatch-repair proficient; MSH, mutS homolog; MLH1, mutL homolog 1; PMS1, PMS1 Homolog 2; SNV, single-nucleotide variations; indels, insertions-deletions; TMB, tumor-mutational burden; HRD, homologous-recombination deficiency; DSB, double-strand break; BRCA, breast cancer gene; CNAs, copy number alterations; CIN, chromosomal instability; mtDNA, mitochondrial DNA; cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; IRF, interferon regulatory factor; NF-κB, nuclear factor κB; ISG, interferon stimulated genes; CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand; CD, cluster of differentiation; NK, natural killer; DC, dendritic cell; TNF, tumor necrosis factor; IL, interleukin; IFN, interferon; IFNR, interferon receptor; JAK1, janus kinase 1; STAT, signal transducer and activator of transcription; P, phosphorylation.
Individuals with germline mutations in one or more MMR proteins (i.e. MLH1, MSH2, MSH6, PMS2), known as Lynch Syndrome (LS) patients, have a higher risk for developing early-onset cancers in multiple tissues (19–21). This indicates that loss of MMR functionality renders cells susceptible to malignant transformation (22, 23). In addition to LS patients, MMR is also frequently affected in sporadic (non-hereditary) cancers due to somatic mutations or loss of expression of one or more MMR proteins (24). The most common cause of MMR deficiency in human cancers is epigenetic silencing of the MLH1 promoter (24–27). Sporadic MMR-deficient (MMRd) tumors are most commonly colorectal carcinoma (CRC) [15% of all CRC (24)] and endometrial carcinoma (EC) [30% of all EC (28)] but can also arise in several other tissue types including stomach, brain (mainly glioblastoma), ovarian, and pancreas, among others (29–31). The reasons why certain tissues develop MMRd tumors more frequently than others are yet unknown.
Loss of function of one or more proteins of the MMR system results in failure to repair replication errors and thus persistence of mutations throughout the genome, particularly in regions of repetitive DNA sequences, known as microsatellite (MS) regions (32, 33). Errors in MS regions most commonly arise from polymerase slippage. This phenomenon is known as microsatellite instability (MSI) and is a hallmark of MMRd tumors. When MSI occurs in coding regions, it can lead to alterations in open reading frames, yielding functionally inactive proteins, including proto-oncogenes and tumor suppressor genes (31, 34). In addition to MSI, MMRd results in a high rate of single nucleotide variation (SNVs) and large numbers of frameshifts that result in indels. Therefore, MMRd cancers are typically considered hypermutated tumors that display a substantial tumor mutational burden (TMB) and a high number of predicted neoantigens (24, 31, 35, 36) (Figure 1A).
Besides MMRd cancers, there is a subset of tumors that harbor mutations in the exonuclease domain of the catalytic subunit of the DNA polymerase epsilon (POLE) or the polymerase delta (POLD1), resulting in loss or defective proofreading (37, 38). These tumors can also arise from a germline or somatic mutation in POLD1 or POLE genes, with POLE mutations in 1%-10% of all CRC and EC cases (24, 38–40). Even though these tumors have relatively low MSI rates and display different MSI signatures than MMRd (41), POLE-mutant (POLE-mut) tumors have dramatically higher rates of SNVs and are therefore considered ultra-mutated tumors (37, 42). While MSI is high in MMRd tumors and SNV rates are high in both groups, in general, MMRd and POLE-mut tumors display relatively stable karyotypes and low numbers of copy number alterations (CNAs) (43).
2.1.2 Defects in homologous recombinationHR is the mechanism primarily responsible for repairing double-strand breaks (DSBs) in DNA during the S and G2 phases of the cell cycle. Upon recognition of DSBs, the 5’ end of the break is resected, creating a ssDNA region that serves as a template for the repair. The exposed ssDNA regions are protected from degradation. BRCA1/BRCA2 mediate recruitment at the DSB site and facilitate the repair. After strand invasion by RAD51, a complex DNA structure is formed (D-loop) followed by DNA synthesis. The newly synthesized DNA can be directly ligated to the original DNA, or alternatively, a holiday junction (HJ) is formed that needs to be further resolved or dissolved by specific proteins to complete the repair (44).
Germline mutations in HR genes including BRCA1 or BRCA2 predispose to early-onset breast and ovarian cancer (45), indicating that the genomic instability caused by HR defects drives malignant transformation. In addition, somatic alterations in BRCA1/2 and other HR-related genes such as ATM or RAD51 are prevalent among several cancer types including ovarian, breast, pancreas, and prostate, among others (46). A defective or compromised HR system leads to unrepaired DSBs, accumulation of DNA lesions, and collapsed replication forks resulting in complex genomic rearrangements and increased susceptibility to accumulation of mutations. In line with this, the genomes of HR-deficient (HRD) cancers are generally characterized by complex CNAs and are highly aneuploid, but may also display specific base substitutions and indels, thus yielding an intermediate TMB (46–49). In fact, cancers with mutations in HR genes have a higher TMB and higher number of predicted neoantigens in comparison to their HR-proficient counterparts (48, 49) (Figure 1B).
2.2 Chromosomal instabilityCIN is defined as persisting errors in chromosome segregation during mitosis, leading to numerical and/or structural chromosomal abnormalities in the resulting daughter cells (50). Aneuploidy is the result of CIN, and it is a state in which cells harbor alterations in the chromosomes. CIN can yield numerical aneuploidy i.e. an abnormal number of whole chromosomes, as well as structural aneuploidy, when chromosome fragments display CNAs due to unbalanced translocations, inversions, or deletions (50) (Figure 1C). Faithful chromosome segregation relies on the structural integrity of the microtubule spindle machinery and the spindle assembly checkpoint (SAC). The various processes that can lead to aneuploidy have been extensively described elsewhere and include but are not limited to centrosome amplifications, telomere dysfunction, deregulation of genes involved in the SAC, loss of cohesion, altered microtubule polymerization rates or abnormal kinetochore-microtubule dynamics (51).
Numerical and structural aneuploidies will lead to unbalanced gene expression, thus disrupting normal cellular processes, and being detrimental to the viability of healthy cells. Furthermore, CIN and aneuploidy promote proteotoxic stress, replication stress, and increased DNA damage, which often leads to cell cycle arrest or cell death (52). Even though CIN and aneuploidy are not well tolerated in healthy tissues (53, 54), over 80% of all human solid tumors display chromosomal abnormalities (55). Furthermore, CIN and/or aneuploidy are associated with (multi) therapy resistance (56–58), immune evasion (59–61), metastasis (62, 63), and thus an overall poor patient prognosis (64, 65). This can likely be explained by the fact that ongoing CIN will promote the generation of new karyotypes during tumorigenesis, thus driving cancer cell evolution and intratumor karyotype heterogeneity (66). Therefore, tumors with high CIN rates will have a large variety of distinct karyotypes and upon (new) selective pressure, cells with specific chromosome combinations may hold a survival advantage and can be selected for.
3 Inflammatory signaling in genomically unstable cancersDNA is normally localized in the nucleus and mitochondria. Genomic instability can lead to DNA being exposed into the cytoplasm for instance as a result of stalled replication (67), mitochondrial damage (68), or by mis-segregated chromosomes and chromosome fragments that yield micronuclei (69). Micronuclei are prone to rupture, eventually exposing the genomic DNA to the cytoplasm (70). Cells are equipped with pattern recognition receptors (PRR) that sense loss of cellular homeostasis, including mislocalized or aberrant DNA and RNA structures. Cytoplasmic nucleic acid sensors are PRRs that signal foreign and host-derived DNA and RNA and when activated, initiate cell autonomous and cell extrinsic defense mechanisms (71).
A well-known cytoplasmic DNA sensor is the cyclic GMP-AMP synthase (cGAS), which recognizes and responds to cytosolic double-stranded DNA (dsDNA) in a DNA-sequence independent manner, serving as a ubiquitous DNA sensor (72, 73). Besides dsDNA originating from the nucleus, other forms of nucleic acid are also able to activate cGAS such as oxidized self-DNA (74), mitochondrial DNA (mtDNA) (75, 76) and, as more recently shown, DNA-RNA hybrids (77). These and other mislocalized nucleic acid structures such as endogenous dsRNA, ssRNA, or mitochondria dsRNA can also be recognized by other cytosolic sensors including RIG-I, MDA5, AIM2, IFI16, and TLRs, among others [reviewed elsewhere (71)]. Here, we will focus on the role of cGAS/STING activation in cancers with high genomic instability, including tumors with DNA repair defects and tumors displaying CIN/aneuploidy.
3.1 The cGAS-STING pathwaycGAS binding to cytoplasmic dsDNA leads to its enzymatic activation and subsequent production of cGAMP, a second messenger molecule and a potent agonist of Stimulator of Interferon Genes (STING) (78). Activated STING recruits TANK-binding kinase 1 (TBK1) to initiate further downstream signaling that ultimately leads to translocation of interferon regulatory factor 3 (IRF3) dimers to the nucleus and the transcription of anti-viral-like gene programs such as type I interferons (IFN) as well as pro-apoptotic genes (79) (Figure 1D). Type I IFNs in turn activate interferon-stimulated genes (ISG) in an autocrine and paracrine manner, including genes in the janus kinase (JAK)/Signal transducer and activator of transcription (STAT) pathway. Whereas JAK/STAT1 signaling plays a key role in the induction of cell death and anti-tumor immunity, JAK/STAT3 signaling has anti-apoptotic effects and promotes cell growth (80). Parallel to IRF3, STING can also activate both the canonical (RELA-p50) and non-canonical (RELB-p52) Nuclear Factor-κB (NF-κB) to induce other inflammatory gene programs (79, 81) (Figure 1D). These signaling cascades do not operate independently but rather constitute a complex signaling network displaying multiple levels of crosstalk and feedback control that result in a pleiotropic response (81). Yet, the magnitude of this response, its dynamics, and the interconnections between these pathways are still poorly understood.
Over the last decade, the cGAS/STING pathway has received significant attention in the cancer field as it plays a pivotal role in mounting anti-tumor responses both in a tumor cell autonomous and non-autonomous manner. Chronic activation of cGAS/STING can result in the secretion of soluble factors collectively known as the senescence-associated secretory phenotype (SASP), which can restrict malignant growth (82–84). Additionally, cGAS/STING-induced pro-inflammatory chemokines and cytokines have a wide range of immune-stimulatory effects shown to induce potent anti-tumor responses (85). Importantly, intrinsic tumor STING expression helps in immune-mediated control of metastatic quiescent cancer cells (86). The cGAS/STING pathway can also coordinate multicellular immune responses through extracellular signaling of cGAMP, amplifying pro-inflammatory signaling (87). cGAMP intracellular communication can be mediated by cell-to-cell junctions that directly connect the cytosol of adjacent cells or by cGAMP-containing exosomes secreted in the extracellular space that can be taken up by recipient cells. In the context of anti-tumor immunity, cancer-derived cGAMP or cancer-derived DNA have been shown to activate dendritic cells and macrophages, which in turn respond by producing type I IFN to enhance CD8+ T cell anti-tumor activity (88–90). Similarly, release of damaged DNA resulting from telomere stress also results in enhanced immune responses (91). This is in line with the observation that NK cell cytotoxicity against tumor cells requires STING expression by host cells and cGAS expression by cancer cells (90). Exploring whether extracellular cGAS/cGAMP signaling is particularly relevant in tumor cells with high genomic instability remains to be investigated.
Conversely, other work has shown a tumor-promoting role for the cGAS/STING pathway. In preclinical models, chronic inflammation induced by 7,12-dimethylbenz(a)anthracene (DMBA) was shown to promote skin tumorigenesis in a STING-dependent manner through IL-6 expression (92). Further studies have shown that cGAS/STING signaling can promote tumor cell survival in an autocrine and paracrine manner (93, 94). How these opposite roles are mechanistically regulated remains unclear, but their dynamics likely determine the ultimate output of cGAS/STING, thereby being very context dependent.
The inflammatory consequences and anti- and pro-tumor effects triggered by sensing of cytosolic DNA in tumors with defects in DNA repair and chromosome imbalances are described in more detail below.
3.1.1 cGAS/STING-driven inflammatory signaling by deficient MMRRecent work has shown that MMRd human tumors, as well as MMR-deficient engineered cell lines, display enhanced type I and type II IFN signatures in comparison to their MMR-proficient (MMRp) counterparts and that this is mediated by activation of the cGAS/STING pathway (95–97). Mechanistically, cells with an inactivated MLH1 gene accumulate cytosolic DNA by dysregulation of MLH1-dependent EXO1 function, which causes unrestrained DNA hyperexcision and aberrant DNA breaks (95). As a result, MLH1 KO cells accumulate cytoplasmic DNA, which eventually results in the expression of ISG15 and IFN-β in a cGAS and STING-dependent manner (97). Further in vitro and in vivo studies have demonstrated that cancer-cell intrinsic cGAS activation and subsequent type I IFN expression is necessary for optimal CD8+ T cell priming. While defects in MMR reduced in vivo tumor growth in comparison to wild-type tumor cells, concomitant deletion of tumor cGAS, STING, or IFNAR1 rescued these growth defects, indicating a key role of cGAS/STING-type I IFN axis in immune surveillance (97). These and other studies have reported the importance of type I IFN signaling in dendritic cell (DC) -T cell interactions (98–101) (Figure 1E).
However, type I IFN is not the only factor produced upon cGAS engagement. CCL5 and CXCL10 chemokines are also upregulated in response to cGAS/STING. Indeed, anti-tumor immunity in MMRd cancers also depends on the magnitude of cGAS/STING activation and subsequent expression of CCL5 and CXCL10 in the tumor microenvironment (TME), which influences CD8+ T and NK cell infiltration (96, 102) (Figure 1E). In vivo, blockade of the CXCR3-CXCL10 interaction with monoclonal antibodies dampened the infiltration of immune cells into the tumors and accelerated tumor growth (96). In comparison to MMRp CRCs, gene and protein expression levels of both cGAS and STING are significantly higher in MMRd, and such tumors display higher CXCL10, CCL5, and CD8+ T effector gene signatures (103, 104). In fact, both cGAS and STING expression correlates with an increase in CD8+ but not CD4+ T cell numbers (104). Although a similar phenotype with accumulation of cytoplasmic DNA and enhanced expression of type I IFN, ISG15, CXCL10, and CCL5 has been shown for MSH2 KO cell lines (96), the mechanism of cGAS activation upon MSH2 deficiency remains to be determined. Altogether, these data underscore the importance of the cGAS/STING-driven inflammatory phenotype in MMRd tumors for optimal anti-tumor immunity.
3.1.2 cGAS/STING in cancers with HR defectsIn the context of HR, loss, inhibition or mutations in BRCA1/2 lead to the generation of cGAS-positive micronuclei and induction of cGAS/STING signaling which results in the expression pro-inflammatory type I and type II IFNs, NF-κB dependent TNFα activation, as well as expression of CXCL10, CXCL9, CCL5, IL-6, and IL-8 (105–109), extensively reviewed elsewhere (110) (Figure 1F). Similar effects are observed in cells with alterations in other HR genes, defective DDR, or upon DNA-damaging agents (109, 111, 112) (Figure 1F). For instance, defects in ATM also result in cytoplasmic accumulation of STING, its subsequent activation, and the initiation of a type I IFN response (113).
In breast cancer, higher cGAS/STING scores are associated with higher genomic instability, which in turn correlate with overall higher infiltration of immune cells (114). BRCA1/2 mutations are indeed often associated with an increased number of tumor-infiltrating lymphocytes in different cancer types (108, 109, 115, 116) and tumor-associated inflammation in BRCA1-mutant breast cancer has significant positive prognostic value (108, 117). In fact, tumors with HR mutations are generally associated with a better patient prognosis than their wild-type counterparts, beyond BRCA mutations (118). Therapeutically, HR deficiency sensitizes tumors to DNA-damaging agents and poly(ADP-ribose) polymerase (PARP) inhibitors. Studies using mouse models of breast and ovarian cancer have revealed that the efficacy of PARP inhibition in BRCA-deficient tumors depends on the activation of the cGAS/STING pathway. Mechanistically, PARP inhibitors increase expression of type I IFN in the TME in a cGAS/STING-dependent manner, which in turn mediates recruitment and activation of effector T cells (119, 120) and NK cells while reducing the number of myeloid cells (121) (Figure 1F). Overall, cGAS/STING activation appears to be relevant for enhancing anti-tumor immunity in the context of HRD cancers both at baseline and upon DNA-damaging therapies.
3.1.3 cGAS/STING in tumors displaying CIN or aneuploidyChromosome mis-segregation events during mitosis can lead whole chromosome and chromosome fragments to end up in the cytoplasm as micronuclei. Rupture of the micronucleus membrane exposes the DNA in the cytoplasm, activating the cGAS/STING pathway (122, 123). Mackenzie and colleagues demonstrated that cGAS quickly localizes to such micronuclei and that ISG genes including CCL5 and CXCL10 are induced almost exclusively in micronuclei-positive cells (69). Indeed, drug-induced CIN triggers type I IFN, canonical and non-canonical NF-κB and eventually STAT1 and STAT3 signaling in an autocrine and paracrine manner (62, 93, 124, 125), extensively reviewed elsewhere (126) (Figure 1G). While micronuclei appear to be an important hub for cGAS activation, recent work suggests that the formation of chromatin bridges prior to micronucleus formation is required to activate the cGAS/STING pathway (127).
In addition to cells exhibiting ongoing CIN, stable aneuploid cells also display an inflammatory phenotype. In contrast to euploid cells, cells with trisomy 21 show a constitutive IFN signature and enhanced ISG response upon IFN stimulation (128). In fact, there seems to be a general inflammatory response to the presence of any extra chromosome as the cGAS/STING/TBK1/IRF3/STAT1 axis was found to be constitutively active in cells with any trisomy, presumably due to accumulation of cytoplasmic dsDNA (129). Furthermore, cells with complex aneuploid karyotypes show transcriptome signatures that include type I IFN response, allograft rejection, antigen processing and presentation as well as activation of NF-κB signaling (6). Another inflammatory characteristic of CIN and aneuploid cells is increased expression of SASP-like cytokines, such as IL-1β, CXCL8, CCL2, CCL27, and TNFs (130). Together these pathways may sustain a chronic inflammatory phenotype, potentially resulting from cGAS/STING engagement, which might aid in the clearance of aneuploid and cells displaying CIN by the immune system.
Conversely, recent work has shown that chronic activation of cGAS/STING favors tumor growth, specifically in the context of cancers displaying high levels of CIN. For instance, cGAS/STING was shown to contribute to increased cancer cell survival through autocrine IL-6/STAT3 signaling in cells with induced CIN phenotypes (93) (Figure 1G). Furthermore, cytosolic DNA was shown to promote metastasis in a cell-autonomous manner via STING/non-canonical NF-κB activation (62) as well as in a non-autonomous cell manner via STING-dependent modulation of the immune system (131). Interestingly, cGAS/STING can lead to the activation of non-canonical NF-κB signaling without altering IFN signaling, indicating that cancer cells can eschew type I IFN signaling while benefiting from cGAS/STING-induced pro-tumor inflammation (62, 131). Collectively, this work demonstrates that cGAS/STING activation has both anti and pro-tumorigenic effects in a context-dependent manner. Acute cGAS/STING activation may lead to cell death, cell cycle arrest, and immune surveillance, whereas chronic cGAS/STING stimulation may ultimately promote survival and initiation of metastasis.
3.1.4 Mitochondrial genomic instability and cGAS/STING signalingMtDNA is also replicated over cell divisions and mitochondria have multiple replication and repair mechanisms to maintain mtDNA integrity (132–135). Similar to nuclear DNA, defects in mtDNA repair mechanisms have been associated with increased malignant potential (136). Furthermore, many cancer types are thought to harbor driver mutations in the mtDNA (137), highlighting the role of mitochondrial genomic instability in tumorigenesis. Beyond nucleotide mutations, mtDNA is vulnerable to damage due to its proximity to the mitochondrial electron transport chain, where reactive oxygen species (ROS) are generated as byproducts of cellular respiration. Under cellular stress, mitochondrial damage or mitochondrial dysfunction, mtDNA or oxidized mtDNA (ox-mtDNA) can exit the mitochondria and activate cGAS/STING. This triggers inflammatory signaling including expression of ISGs and the production of type I IFNs and chemokines such as CXCL10 (68, 138, 139), which may have intrinsic and extrinsic effects. Additionally, mtDNA has been shown to contribute to the SASP via activation of the cGAS/STING (140) and extracellular mtDNA release by senescent cells contributes to an immunosuppressive TME (141).
It is important to notice that, beyond cGAS/STING, mtDNA (and mtRNA) can activate a wide range of other inflammation pathways including NLRP3 inflammasome, RIG-I, TLR9, ZBP1 or IFI16, described in more detail elsewhere (142). Despite this evidence, the specific consequences of mtDNA-driven inflammation within tumors, as well as its impact on the overall immune environment and response to immunotherapy are yet to be fully understood. Furthermore, it would be interesting to investigate whether tumors with high nuclear genomic instability (e.g. CIN) also exhibit increased mitochondrial genomic instability.
Next to MMRd, HRD or CIN, it would be interesting to understand how other forms of genomic damage or genomic alterations such as centromere defects (143), DNA damage at telomeres (144), chromotripsis or toroidal nuclei affect cGAS/STING-driven inflammation and immune responses and how these pathways intersect with each other.
4 Immunogenicity of genomically unstable cancersTumor immunogenicity is the ability of tumor cells to induce a strong immune response, which can greatly vary between cancer types and individuals. Key determinants of tumor immunogenicity include but are not limited to the specific oncogenic drivers (145–147), the TMB (7) and/or the intrinsic inflammatory phenotype of the tumor cells (148). How the different types of genomic instability trigger inflammatory signaling and immune surveillance and how these pathways are intertwined in tumors with high genomic instability is described in more detail below.
4.1 Cancers with MMR defectsThe genomic instability resulting from MMR deficiency significantly impacts the TME, the progression of the disease and the response to (immuno)therapy. In comparison to their proficient counterparts, MMRd tumors have been consistently reported to have an inflamed microenvironment with high infiltration of cytotoxic immune cells, which is correlated with an overall better prognosis and, importantly, better response to immunotherapy (149–151).
In the last decade, transcriptional profiles of bulk tumor samples and single cells from large cohorts of CRCs allowed for the classification of these tumors in 4 consensus molecular subgroups (CMS) based on their cellular composition (43, 152). The first CMS was enriched for MMRd/MSI-high tumors displaying high TMB and low somatic CNAs (SCNAs). In comparison to the 3 other CMS, the MMRd-enriched subgroup shows significantly higher infiltration scores of cytotoxic lymphocytes and Th1 cells, low infiltration of regulatory T cells, and high expression of genes encoding for cytotoxic T cell attracting chemokines, Th1 cytokines, and other cytokines and chemokines involved in anti-tumor immunity such as IFNs, CXCL13 and IL15 (43, 152). These findings were further substantiated using the CIBERSORT algorithm in a more recent study (153) and observed in other MMRd tumor types, including EC (39, 154, 155). Furthermore, MMRd tumors have been shown to generate systemic robust immune responses (156). The diverse factors contributing to increased immunogenicity and immune activation in MMRd tumors are reviewed below.
4.1.1 TMB, neoantigens and CD8+ T cellsMMRd tumors accumulate many mutations in the DNA sequence, quantified as the TMB (157). This may result in the production of mutated proteins that can eventually be presented as (neo)antigens loaded in the Major Histocompatibility Class-I (MHC-I) complex. MHC-I complexes are expressed on the surface of all nucleated cells and continuously present the host proteome in the form of peptides to CD8+ T cells, thus playing a critical role in the adaptive immune system (158). CD8+ T cells are MHC-I-restricted, and therefore can only recognize peptides loaded in the MHC-I. The MHC-I-peptide complexes are scanned by CD8+ T cells, which mount potent, specific, and long-lasting immune responses when non-self-peptides are identified (159). Several studies have shown a very strong positive correlation between TMB, CD3+, or CD8+ T cell infiltration and favorable prognosis in many cancer types, particularly in MMRd ones (103, 160–163). In fact, the number of frameshift mutations and the number of predicted neoantigens correlates with the density of infiltrating CD8+ T cells and lymphocyte score, respectively (163, 164) (Figure 2). Not only do MMRd tumors contain higher numbers of tumor-infiltrating T cells, but these T cells also have higher expression of activation and cytotoxic markers such as IL-2Rα (165), granzyme B (165, 166), perforin (167), PD-1 and IFN-γ (153, 162, 168, 169). Indeed, single-cell RNA-sequencing experiments revealed that the T cell compartment is the key difference between the immune composition of MMRd and MMRp CRCs (170). In line with previous observations, tumor infiltrating T cells in MMRd tumors exhibited gene signatures related to cytotoxicity (granulysin, granzymes, and perforin) as well as activation (PD-1) consistent with chronic stimulation. Furthermore, MMRd tumors contain abundant T cells with a strong CXCL13+ signature, suggesting effector tumor-specific T cells (170, 171). Similarly, MMRd ECs are enriched in CD8+ T cells expressing high levels of PD-1, CD39, TIM-3 and CXCL13, which define a population of tumor-reactive T cells that was also positively correlated with the level of the TMB (160) (Figure 2).
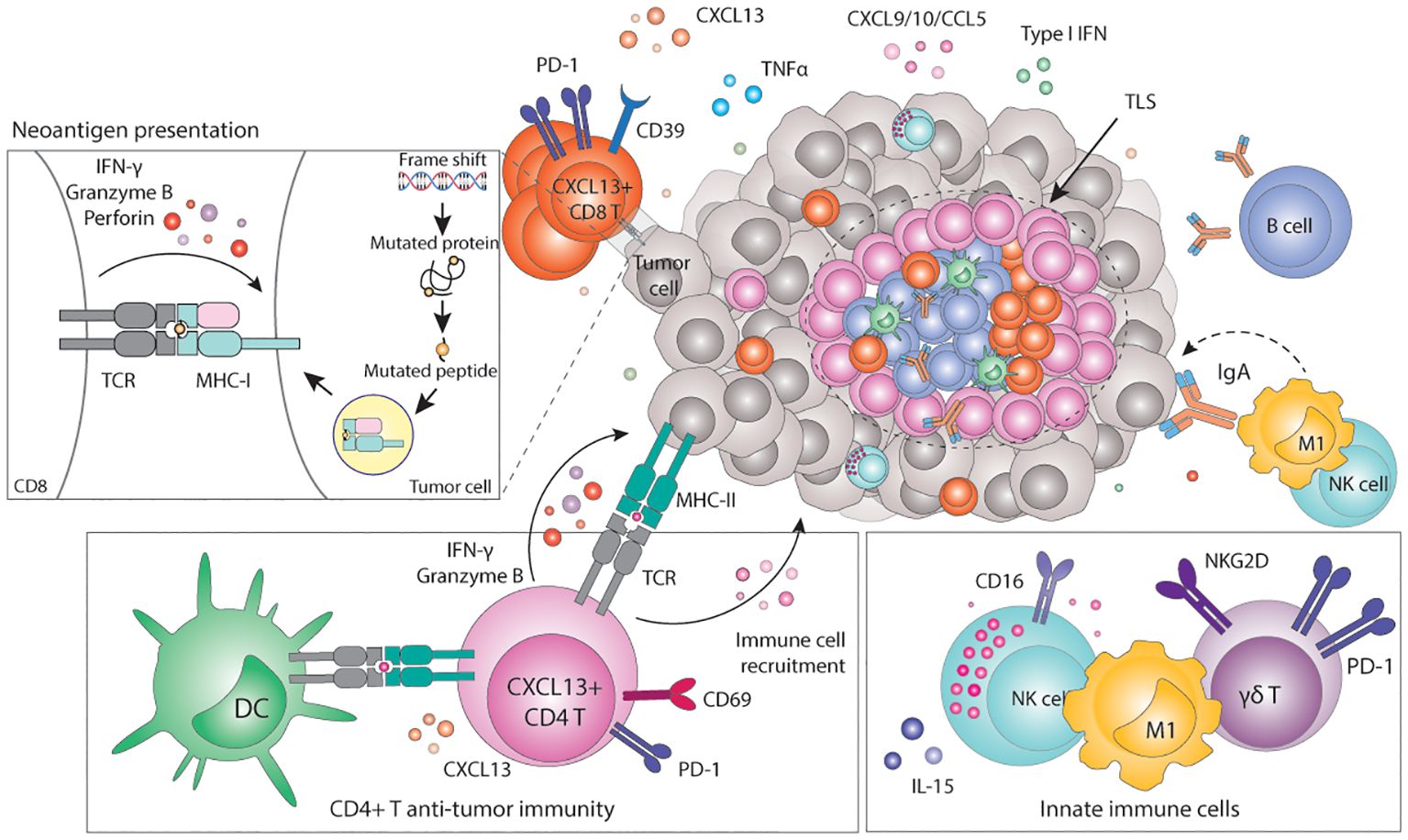
Figure 2. Immunogenicity of MMRd tumors. Frameshifts and SNV eventually result in the expression of neoantigens loaded in the MHC-I complex. Recognition of neoantigens activates CD8+ T cells and subsequent expression of cytotoxic molecules including granzyme B, IFN-γ or perforin, leading to the elimination of tumor cells. Tumor cells can also express neoantigens loaded on the MHC-II, which can be recognized by CD4+ T cells and initiate a cytotoxic response. In parallel, CD4+ T cells can also express pro-inflammatory cytokines and chemokines to recruit myeloid and NK cells that contribute to enhance anti-tumor immunity. Innate γδ T cells can target tumor cells via the NKG2D ligand-receptor interaction. High TMB correlates with the presence of TLSs in the tumor, which are highly organized hubs of immune cells that shape both adaptive and humoral immune responses. In particular, TLSs contain B-cell producing antibodies that may bind tumor antigens and trigger antibody-dependent cytotoxicity (ADCC). MHC, Major histocompatibility complex; TCR, T cell receptor; CD, cluster of differentiation; PD-1, programmed-death 1; TLS, tertiary lymphoid structure; Ig, immunoglobulin; CXCL, C-X-C motif chemokine ligand; CCL, C-C motif chemokine ligand; TNF, tumor necrosis factor; IFN, interferon; M1, macrophage type 1; NK, natural killer; IL, interleukin; NKG2D, natural killer group 2 D.
The current dogma is that the high TMB drives the inflammatory and immune activated phenotype. Indeed, pre-clinical studies have shown that defects in DNA repair trigger neoantigen generation and promote immune surveillance of tumors in vivo in a CD8-dependent manner (7, 150). Moreover, the degree of TMB within MMRd tumors affects their growth rate and response to ICB (150). More interestingly, in preclinical MMR heterogeneous tumors, the immunogenicity of MMRd cells was shown to drive the elimination of MMRp cells within the same local microenvironment (172). Clinically, TMB or surrogate markers such as MMRd are often one of the strongest predictors of response to ICB across cancer types (151, 173–175) and the degree of TMB and MSI status (MSI-high vs MSI-low) predicts long-term benefit to ICB (153). However, despite being a relevant factor, increasing evidence suggests that TMB is necessary but not sufficient to fully explain the strong anti-tumor immunity towards cancers with a hypermutated phenotype (176). In studies using murine lung and colon cancer models, MMRd and the resulting high TMB were not sufficient to increase the immunogenicity nor sensitivity of tumors to ICB. Instead, the presence of clonal neoantigens was shown to be more critical for effective T cell responses (177). Furthermore, the fact that cGAS or STING-deficient MMRd cell lines grow faster than the cGAS or STING-proficient MMRd counterparts in immunocompetent mice supports the idea that anti-tumor immunity does not solely rely on the expression of neoantigens (97). These observations underscore the importance of (cGAS/STING) inflammatory signaling and antigen-independent mechanisms of immune recognition and activation in cancers with MMR defects.
4.1.2 CD4+ T cell anti-tumor immunityIn addition to the high infiltration of cytotoxic CD8+ T cells, MMRd tumors also show significant infiltration of CD4+ T cells compared to their MMRp counterparts. Premalignant polyp lesions of LS patients are densely infiltrated by IFN-γ-expressing CD4+ T cells, suggesting a role for CD4+ T cell recruitment and activation early during tumorigenesis (167). Similarly, significant CD4+ T cell infiltration has been observed in MMRd carcinomas, though in variable proportions. These effects also differ per tumor type: while in CRC CD8+ T cells are typically the dominant T cell type, MMRd ECs have higher densities of CD4+ T cells (160). CD4+ T cells more often localize in the invasive front and express higher levels of PD-1 and IFN-γ compared to MMRp tumors (168). Flow cytometry analysis showed enrichment of IFN-β-expressing CD4+ T cells in MMRd CRCs (178). Furthermore, these cells were positive for CXCL13 and co-express high levels of PD-1, CD27, CD39 and Ki67 (178), suggesting tumor reactivity (179). Indeed, PD-1high CD4+ T cells isolated from MMRd ECs co-expressed other activation markers such as CD38, HLA-DR, ICOS, BCL6, CXCL13 and KI67, were proven to be tumor-reactive T cells and correlated with improved prognosis in ECs with high TMB (160). Interestingly, CD4+ T cells can drive strong anti-tumor immunity upon ICB in preclinical models of MMRd tumors with defective or low expression of MHC-I, which cannot be recognized by CD8+ T cells (180, 181). Under these circumstances, CD4+ T cells show high expression of the activation markers PD-1 and CD69, and cytotoxic molecules such as granzyme B (180) (Figure 2). However, the mechanisms of CD4+ T cell mediated control of MMRd MHC-I-deficient tumors remain to be fully elucidated.
Emerging evidence in different models indicates that CD4+ T cells may play a role in anti-tumor immunity, distinct from their conventional function as helpers and regulators of cytotoxic CD8+ T cells (182). A subset of CD4+ T cells can acquire cytolytic function towards MHC-II-expressing tumor cells (183, 184). MHC-II is generally expressed by professional antigen presenting cells (APCs) and is used to present extracellular peptides from pathogens or tumors to CD4+ T cells. In contrast to CD8+ T cells, CD4+ T cells are MHC-II-restricted and therefore, they can only recognize peptides loaded onto the MHC-II complex. It has been reported that, under certain conditions, tumor cells can also express MHC-II. Because of the high TMB of MMRd tumors, it could be that neoantigens are also presented in the context of MHC-II, activating potent CD4+ T cell anti-tumor immunity. Indeed, CD4+ T cells isolated from MMRd ECs showed MHC-II-restricted tumor reactivity to autologous tumor cells (160)(Figure 2). Other work has shown that CD4+ T cells can target tumor cells independently of MHC-II by mobilizing or activating myeloid cells and NK cells (182, 185, 186). Overall, this data suggests that CD4+ T cells have a prominent cytotoxic role in anti-tumor immunity in MMRd tumors (including MHC-I proficient ones) more than previously anticipated.
4.1.3 Innate immune cellsAs opposed to the adaptive immune system, the role of innate immunity in MMRd tumors remains largely unexplored. Increased infiltration of innate γδ-like T cells has been reported in some MMRd CRC cohorts (170, 187) and these cells express higher levels of PD-1 in comparison to MMRp tumors (170). Analysis of MMRd tumors treated with a combination of anti-PD-1 and anti-CTLA-4 showed a role for γδ-T cells in targeting MMRd tumor cells with defective antigen presentation potentially via de NKG2D/NKG2DL ligand-receptor interaction (188). Other studies have found a positive correlation between MMRd status and infiltration of both macrophages and NK cells (153, 187). Activated NK cell gene signatures were also enriched in MMRd tumors (153) and circulating CD16+ NK cells showed strong upregulation of cytotoxicity and activation genes in EC MMRd patients responding to ICB (156). In comparison to non-responders, NK cells from responding patients express higher levels of granzyme A, EOMES, and TNF-mediated signaling molecules and were associated with longer survival (156). Anti-tumor M1-like macrophages were shown to be predominant in MMRd tumors and had significant prognostic value (187). A more recent study showed that whereas the abundance of monocytes and macrophages is similar between MMRp and MMRd CRC, macrophages from MMRd tumors express more inflammatory factors, chemokines and immune-activating alarmins than their MMRp counterparts (170), potentially contributing to an immunoreactive microenvironment. However, data remains scarce and heterogeneous and the precise functional contribution of innate immune cells in anti-tumor immunity and response to immunotherapy in the context of MMRd tumors remains to be elucidated.
4.1.4 Tertiary lymphoid structures (TLSs) and B cellsAn interesting observation is the positive correlation between neoantigen burden, TMB, and the presence of tertiary lymphoid structures (TLSs) (189). TLSs are highly organized ectopic lymphoid structures, mainly comprised of B and T cells, that provide niches for multiple crosstalk between immune cells. TLSs are thought to shape antigen-specific immune responses, clonal expansion, and increase cytokine-mediated signaling (190, 191). The presence of TLSs is a prognostic factor in several cancer types (192–199), as it is associated with a protective immunity and correlates with favorable outcomes both in primary and metastatic disease (190, 195, 197, 198, 200, 201).
Using a TLS gene signature, Lin and colleagues proposed that neoantigen load and the total TMB correlate with the presence of TLSs in multiple cancer types, including ECs (189). In line with these findings, TLSs were more frequent in MMRd and POLE-mut ECs, and the presence of 1 or more TLSs in these tumor types was a beneficial prognostic value (189, 202). While the mechanism underlying this phenomenon is thus far poorly understood, a contributing factor seems to be the increased presence of B cells and CXCL13-producing T cells in high TMB tumors (203). CXCL13 is a B-cell chemoattractant involved in lymphoid neogenesis and B cell differentiation (204). Workel and colleagues showed that CXCL13+ T cells are enriched in tumors with a high TMB, which correlates with the presence of TLSs (203). In agreement with this, MMRd CRCs display strong TLS signatures and have a significant presence of CXCL13-expressing T cells throughout the tumor and CXCL13-expressing follicular DCs in TLSs (43, 170). Increased presence of TLSs in tumors with a high TMB may also help improve humoral responses. Indeed, compared to other EC subtypes, only MMRd ECs showed an increased abundance of B cell-derived IgA antibodies in the TME. These antibodies were further shown to increase the expression of immunostimulatory cytokines such as TNF and IFN, overall increasing anti-tumor immune responses (199)(Figure 2). In other cancer types, IgA antibodies bind polymeric IgA receptors on tumor cells, which in turn enhances tumor targeting by myeloid and T cells (205). Nevertheless, how exactly the TMB instigates immune cells to drive the formation of TLSs and how this contributes to improved survival remains to be fully elucidated.
4.2 Cancers displaying CIN and aneuploidyThe relationship between cancers displaying CIN and the immune system appears to be very complex. Classification of CRCs based on transcriptional profiles showed a subgroup characterized by high numbers of CNAs (43, 152). Despite the high genomic instability, this subgroup appears to be immunologically “cold” in comparison to the other subtypes. A similar trend is observed in EC, where the TP53 mutant subgroup is characterized by high CNAs, very low immunogenicity and poorer survival outcomes (39, 206). The observation that tumors with low CNAs present a more immune active profile and that high CIN/aneuploid tumors exhibit features of immune exclusion has been made in preclinical models (207) and other cancer types (208). Nevertheless, the reasons for this phenomenon remain to be fully understood.
An interesting study by William and colleagues compared the immune differences between pre-cancerous lesions and later stages of cancer development in HPV- head and neck cancers with major risk SCNAs (209). They found that loss of chromosome 3p, 9p or 17p in pre-cancerous lesions is associated with increased CD3+ T cell infiltration, being trisomy and tetrasomy in chromosome 7 the most strongly associated with overall immune cell density. Interestingly, in later stages of cancer development, the same SCNAs were associated with reduced T cell infiltration, lower cytotoxic activity, and poorer prognosis (209). Furthermore, analysis using TCGA has shown that aneuploid tumors show more features of immune evasion in comparison to non-aneuploid tumors (59, 210). This data demonstrates that CNAs drive a transition from an immune dense to an immune evasive microenvironment over time, but it also suggests they can elicit an immune response, at least in the early stages of tumorigenesis. Subsequent research has shown that acute induction of CIN and/or aneuploidy increases the immunogenicity of tumor cells in vitro and in vivo, whereas chronic CIN and the resulting aneuploidy ultimately leads to immune evasion, which will be described in more detail further below.
Here, we describe the mechanisms by which immune cells can recognize cancer cells mainly with acute induction of aneuploidy/CIN.
4.2.1 Immune recognition of CIN/aneuploid cellsBesides inflammatory signaling, karyotypic abnormalities can also trigger direct immune recognition mediated by membrane-bound proteins. Hyperploid tumor cells as well as tetraploid TP53-/- colon organoids display constitutive endoplasmic reticulum (ER) stress which results in abnormal cell surface exposure of calreticulin (CRT) (211, 212). Interestingly, cells with an extra copy of chromosome 7 did not display ER stress nor abnormal CRT exposure, suggesting that only a major increase in chromosome copy number induces enough ER stress and subsequent CRT surface exposure. Cells with abnormal surface CRT exposure were able to grow in immunocompromised Rag2yc-/- mice, but tumor growth was much slower when injected in immunocompetent mice. Clearance of hyperploid tumors in immunocompetent mice further protected them against rechallenge, indicative of immune memory (211). Indeed, immunosurveillance of hyperploid tumor cells was shown to involve both CD4+ and CD8+ T lymphocytes as well as type I and type II IFN (211) (Figure 3). Even though the precise mechanism for CRT redistribution in the cell surface upon high chromosome numbers is not well described, it is long known that CRT serves as an “eat-me signal” and facilitates phagocytosis of stressed cells by APCs, including macrophages and DCs (213, 214), which could explain the findings. In addition, CRT is known to facilitate MHC-I assembly and folding. Thus, higher levels of CRT may also result in higher MHC-I expression by aneuploid cells, increasing their recognition by T cells (215). Finally, a role for Natural Killer (NK) cells cannot be excluded since the widely expressed NKp46 receptor has been recently shown to recognize CRT as a danger-associated signal to eliminate ER-stressed cells (216). CIN and aneuploidy are also known to induce ER stress, which may also induce CRT exposure, enhancing the immunogenicity of such tumor cells (52, 131).
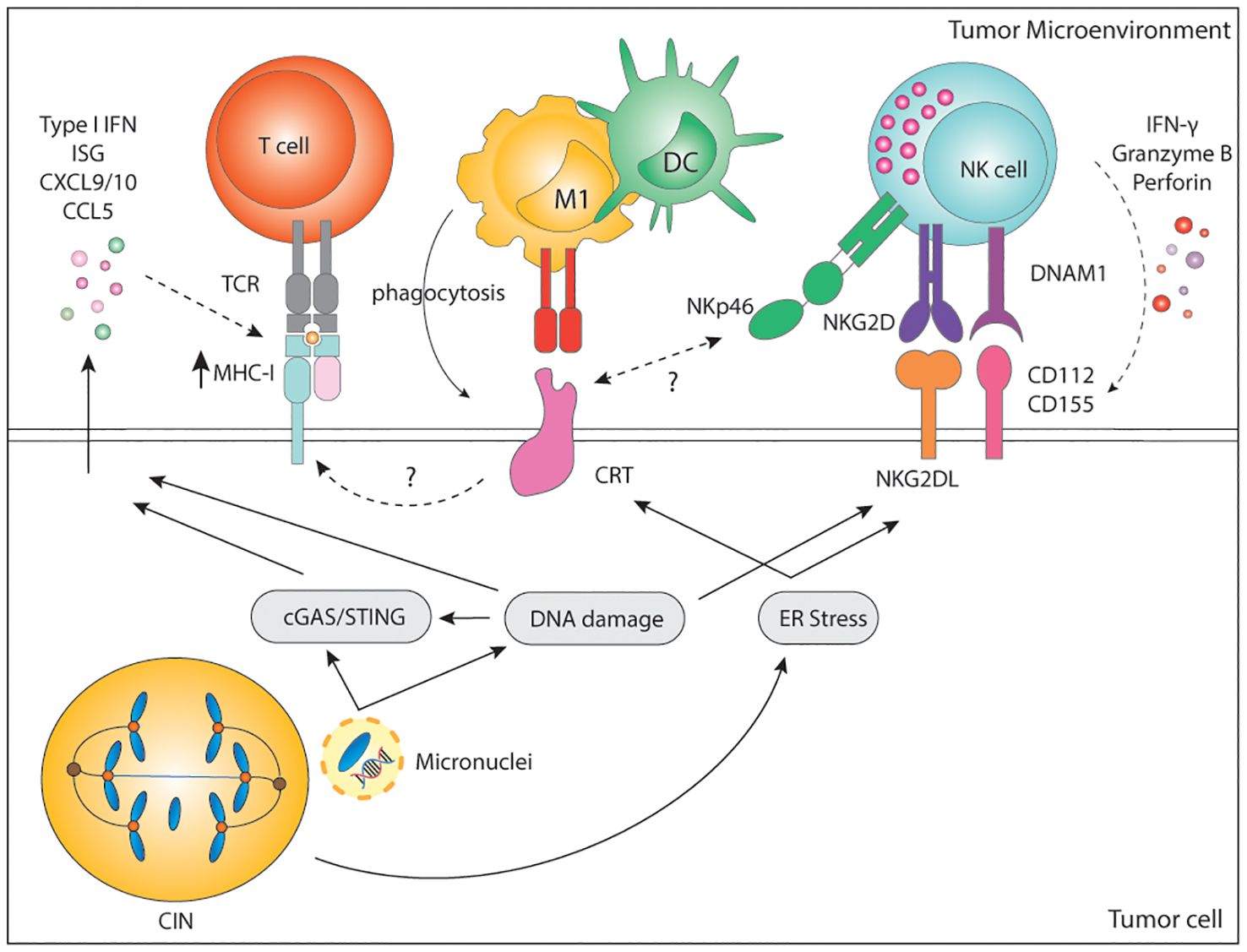
Figure 3. Immune recognition of CIN/aneuploid cells. CIN/aneuploidy trigger a wide range of cellular stressors resulting in the expression of immune activating ligands at the cell surface. CRT is expressed upon ER stress and facilitates phagocytosis by APCs and cytotoxicity by NK cells via the NKp46 receptor. CRT is also known to increase expression of MHC-I for the recognition of stressed cells by CD8+ T cells. In parallel, both DNA damage and ER-stress can upregulate the expression of NKG2D and DNAM-1-ligands, widely known to potently activate NK cells. Finally, secretion of soluble factors by CIN/Aneuploid cells may contribute to immune infiltration, immune activation and increase expression of MHC-I by tumor cells, altogether enhancing their recognition by the immune system. CIN, chromosomal instability; IFN, interferon; ISG, interferon stimulated genes; CXCL, C-X-C motif chemokine ligand; CCL, C-C motif chemokine ligand; MHC, Major histocompatibility complex; TCR, T cell receptor; CD, cluster of differentiation; M1, macrophage type 1; DC, dendritic cell; NK, natural killer; NKG2D, natural killer group 2 D; DNAM-1, DNAX accessory molecule; CRT, calreticulin; ER, endoplasmic reticulum; cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon response cGAMP interactor.
Cultured cell lines with drug-induced aberrant karyotype also show increased immunogenicity in vitro as a result of enhanced expression of stress-related DNAM-1 and NKG2D ligands (6, 217). DNAM-1 (CD226) is broadly expressed by various immune cells, including NK and T cells, epithelial and endothelial cells, and through its ligands, CD112 and CD155, mediates cell-to-cell interactions. The activating NKG2D receptor is mainly expressed in NK cells, γδ-T cells, and CD8+ T cells. NKG2D ligands are a highly diversified MHC-I-like family of self-molecules poorly expressed in healthy cells. Binding of the DNAM-1 or NKG2D ligands to their cognate receptor initiates a signaling cascade that results in activation, IFN-γ, and cytokine release by NK cells. Both DNAM-1 ligands and NKG2D ligands are lowly expressed at baseline but can be strongly induced upon viral infection, stress, senescence, and DNA damage (218, 219). Murine and human NKG2D and DNAM-1 ligands are upregulated by genotoxic stress and stalled DNA replication, both known to activate the DNA damage response (4, 220–222). Moreover, sensing of cytosolic DNA in the form of micronuclei has been shown to upregulate the NKG2D ligand RAE1 in murine lymphoma cells in a STING, TBK1, IRF3 and IFN-dependent manner (223). In human melanomas, there is a significant positive correlation between cGAS expression and human NKG2D ligands ULBP1 and ULBP3 (90). This suggests that not only the cGAS/STING pathway is relevant for the production of soluble factors to alert the innate immune system, but it is also involved in the surface expression of danger-associated signals. Nonetheless, the expression and regulation of DNAM-1 and NKG2D ligands upon genomic instability remains to be fully dissected.
In line with previous findings, drug-induced hyperploid cells upregulate CRT but also DNAM-1 and NKG2D ligands. Primary human NK cells cocultured with drug-induced hyperploid cell lines showed increased proliferation, IFN-γ production, and enhanced cytotoxic capacity (217) (Figure 3). Santaguida and colleagues showed a similar phenotype in cells with drug-induced complex karyotypes, which in vitro were also preferentially killed by the NK92 cell line in comparison to their euploid counterparts (6). NK92 killing of cells with complex karyotypes was further shown to be dependent on both canonical and non-canonical NF-κB but not on type I IFN (224) (Figure 3). However, these latter findings remain to be confirmed in a more physiologically relevant context involving primary NK cells. In summary, these data indicate that acute induction of CIN or a high chromosome content can trigger the expression of activating signals for the rapid elimination of cells.
Similarly, MPS1 inhibition-induced aneuploidy of the B16 cell line was shown to increase overall immune infiltration and to favor macrophage polarization to an anti-cancer M1-like phenotype in vitro and in vivo (225). Further coculture experiments showed that macrophages can suppress the growth of aneuploid/CIN tumor cells in vitro (225
留言 (0)