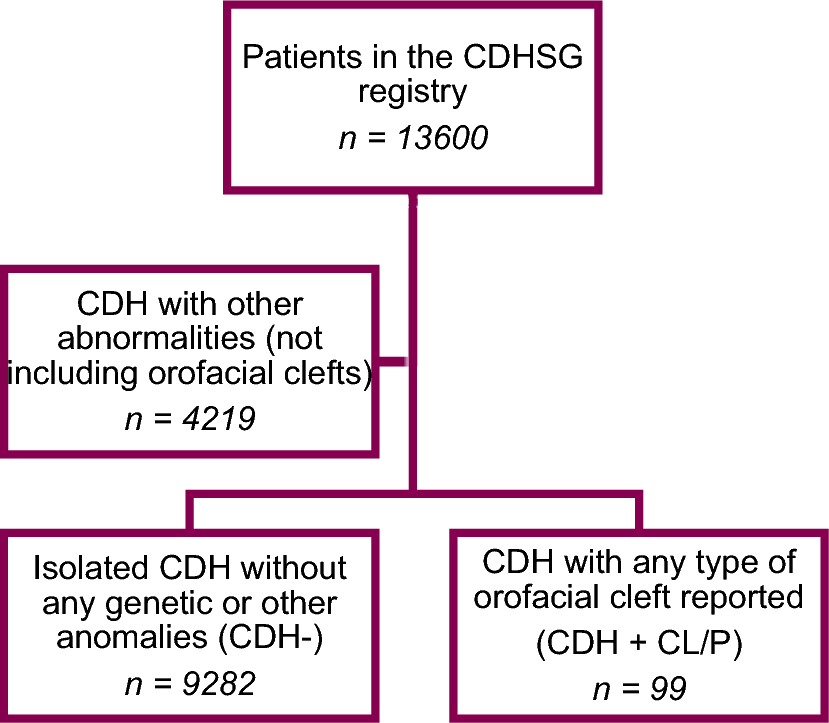
Whether there is an association between CDH and CL/P and what the outcomes for these patients are have not previously been investigated, neither are the genetic and environmental factors contributing to the two malformations fully known. Since these congenital closure defects arise during the same period of intrauterine life, there could be common genetic factors influencing the development of both abnormalities. Trisomy 13, deletion of 8p23.1, and Wolf-Hirschhorn syndrome/4p16.3 deletion were recorded multiple times in CDH+CL/P patients. Based on the registered data, the prevalence of OFC in patients with CDH was estimated to 0.7%, which is markedly higher than the 1–2 per 1000 live births that is reported to be the overall prevalence of CL/P [19]. Hence, it seems that OFCs are somewhat more common in infants with CDH compared to in the non-CDH population. That the CDH+CL/P cohort is generally sicker was shown by higher odds of death early after birth, higher rates of non-repair, and a lower chance of and longer time to discharge. However, after surviving to 7 DOL and after surgical repair, survival rates were not significantly different between groups. Even though the results were not significant, probably due to a low number of CDH+CL/P patients with O/E LHR reported, the severity of lung hypoplasia was shown to be more pronounced in this group, shown by lower O/E LHR values.
Multiple studies have pointed towards a heterogenic etiology for CDH with varying degrees of genetic determinants [10, 23]. This study found 22.2% of the CDH+CL/P patients had registered genetic anomalies, compared to the approximately 10% in all CDH patients that earlier studies have reported, although reports range up to 34% [12,13,14,15,16,17]. This relatively large number points to a higher rate of genetic abnormalities in CDH+CL/P patients. However, it is not the highest number reported, with one possible explanation being that CDH+CL/P patients have another genetic background, to a smaller degree associated to chromosomal abnormalities. That the diagnostic methods have evolved during the study period can also influence the comparison with other studies. Moreover, we cannot exclude the possibility that not all patients entered in the registry have undergone chromosome analysis.
This is the first study to investigate which genetic aberrations occur in patients with both CDH and OFC. Both the deletions (deletion of 8p23.1 and deletion of 4p16.3) and trisomy 13 described in this study have already been reported with CDH, and with CL/P, although to different extents. Trisomy 13 is a chromosomal abnormality with a strong association to CLP [24, 25], and also one of the more common aneuploidies described together with CDH [24], with high antenatal mortality and a survival for live births of less than 10% [26]. Due to the already established associations, the finding is not surprising.
The deletions identified in this study both involve intervals known to be associated with CDH and CL/P. The 8p23.1 deletion is known as a CDH “hot spot”, with an incomplete penetrance of about 50% [27]. There are also reports of the deletion, and the duplication, of 8p23.1 occurring together with an OFC [28, 29]. A gene in this chromosome segment hypothesized to contribute to the development of CDH is GATA-binding protein 4 (GATA4) [10], a transcription factor associated with retinoic acid signaling which is expressed in the developing heart and diaphragm [30]. On the other hand, the 4p16.3 deletion causes Wolf-Hirschhorn syndrome (WHS, MIM: 194190), which is characterized by growth and mental retardation, cardiac anomalies, and dysmorphic craniofacial features [23, 31]. WHS includes an OFC in about 25% of cases [32, 33]. Although not a common feature of the syndrome, CDH has been reported in more than a dozen WHS patients, with multiple genes identified whose haploinsufficiency could contribute to CDH [10, 23, 34]. In summary, the chromosomal abnormalities found in this study correspond well with previous literature on the hypothesized genetic backgrounds of CDH and CL/P.
As far as the authors are aware, the outcomes for patients with CDH and an associated OFC have not been investigated earlier. Although not surprising that outcomes are worse than for patients with isolated CDH, it is noticeable that the OR for death within 7 DOL is almost four times higher for the CDH+CL/P cohort, and that these patients are half as likely to eventually be discharged. The results suggest that, if not operated on at an early age, CDH+CL/P patients have a lower chance of undergoing surgery at all. The significantly higher rates of non-repair and lower rates of ECLS point towards different tendencies in the care given to and resources put into these patients, indicating a sicker and more fragile group. One could speculate that the reduced rate of ECLS indicates a group with a less severe condition but, however, the similarities in defect size and PPHN requiring treatment (as a proxy of severity of the condition) point towards that, presumably, both groups would be needing ECLS to a similar extent. It is therefore plausible that these lower numbers indicate that the infants are presumed beforehand to be too sick to benefit from receiving ECLS.
Strengths and limitations
The main strength of this study is the broad extent of data. Being both multinational and multi-center, the CDHSG registry includes patients from a wide geographical spectrum. We focused on all infants ever to have been reported to the registry, which provides for the largest material possible from the registry. However, the study also has its limitations, mainly due to reporting of data being both limited and varying over time. Some of the parameters, e.g. hernia size, PPHN, and O/E LHR, were introduced in recent versions of the CDHSG reporting form, which means data is missing for a large share of the CDH+CL/P patients. This could decrease the chance of significant results and potentially skew the results, since some studies have found that outcomes have improved for CDH patients over time [8, 35]. As the CDHSG registry does not specify whether associated abnormalities are not present, not detected, or not registered, there is a level of uncertainty regarding this parameter. The methods, protocols, and availability for genetic testing has also changed a lot during the period that the data were collected. Which genetic tests have been performed, if any at all, is not recorded in the current data set, also limiting the interpretation of the results regarding possible genetic factors. As the data collection only comprises the time of in-hospital stay, it should also be noted that it does not consider any events occurring after patients are discharged from the centers.
Clinical applications
This study shines a light on the outcomes for infants with a rare combination of congenital malformations, a combination which has not been researched before. Research on infants with CDH influences guidelines for clinical decision making by pediatricians involved in these patients within both surgery and medicine. Research on infants with CDH is also of importance to the families of patients with CDH as it can help improve prenatal counselling and risk prediction [36]. In this context it is also relevant how the co-occurrence of the two malformations CDH and CL/P affects the expected outcomes. Although not overwhelming, nor previously completely undiscovered, the reported genetic anomalies found in the CDH+CL/P group could be of clinical relevance for parents with children with these genetic anomalies. The next step is to perform comprehensive genetic analysis in these cases, using whole genome sequencing in trio format. This will facilitate the study of monogenic causes in the CDH+CL/P group. With a larger part of the genetic factors elucidated, genetic testing and counseling could influence the advising and the clinical management in the context of prenatally diagnosed CDH, to better help families and individuals with CDH, with or without CL/P.






留言 (0)