
記住我








To test the hypothesis that inflammasome activators promote excess choroidal angiogenesis, we adapted the laser photocoagulation model by applying a single laser burn to wild-type C57BL/6J mice eyes followed immediately by subretinal injection of PBS or Alu RNA, which is an AMD-related inflammasome agonist transcribed from short interspersed nuclear element (SINE) retrotransposons [2, 13] (Fig. 1a). Seven days later, Alu RNA-treated eyes exhibited a dramatic increase in the volume of the CNV lesion compared to saline-treated eyes (Fig. 1b, c). Similarly, subretinal administration of a plasmid expressing Alu RNA (pAlu) with transfection reagent resulted in an increased CNV response compared to the transfection of an empty plasmid (pNull) (Fig. 1d, S1).
Administration of murine SINE B2 RNA, which like Alu RNA is an inflammasome agonist [2, 13], also exacerbated CNV (Fig. 1e, S1). Interestingly, mouse SINE B1 RNA, which is a poorer inflammasome agonist [12], did not significantly affect CNV (Fig. 1e, S1).
Alu RNA can be reverse transcribed by the LINE-1 reverse transcriptase into a complementary DNA (Alu cDNA) which stimulates inflammasome activation, promotes RPE death, and is enriched in the retina of human AMD eyes [16, 17]. Subretinal delivery of synthetic Alu cDNA also increased the volume of CNV lesions (Fig. 1f, S1). To assess whether this angiostimulatory property was unique to SINE-derived oligonucleotides, we tested amyloid-β, another inflammasome agonist that accumulates in human AMD [18,19,20], contributes to spontaneous CNV in a mouse model [21], and promotes inflammasome-dependent RPE death [14, 22]. Subretinal delivery of oligomerized amyloid-β1−40, but not a control peptide (amyloid-β40−1), likewise aggravated CNV (Fig. 1g, S1). These findings indicate that multiple inflammasome agonists of different compositions can amplify the choroidal angiogenic response in the laser injury model.
Fig. 1
Inflammasome agonism immediately following laser injury increases CNV volume. (a) Schematic demonstrating the combined laser CNV and subretinal injection (SRI) model. First, a laser burn is applied to rupture Bruch’s membrane. Subretinal injection is performed immediately following laser injury at the same site, and neovascularization begins to form around day 3. (b) Representative depth-coded 3D projections of laser CNV with SRI of PBS (left) or Alu RNA (right). Dimensions: 633.25 μm x 633.25 μm x 46 μm (c) CNV volumes quantified 7 days after combined laser injury and SRI of Alu RNA (P = 0.0001, Mann-Whitney test. N = 15 per group). (d) CNV volumes quantified 7 days after combined laser injury and in vivo transfection of plasmid-encoded Alu via SRI (P < 0.01, Mann-Whitney test. N = 8 per group). (e) CNV volumes quantified 7 days after combined laser injury and SRI of PBS, B1 (P > 0.99 vs. PBS), or B2 RNA (P = 0.03 vs. PBS, Kruskal-Wallis test. N = 6 (B1), N = 7 (B2)). (f) CNV volumes quantified 7 days after combined laser injury and SRI of Alu cDNA (P = 0.01, Mann-Whitney test. N = 7 (vehicle), N = 7 (Alu cDNA)). (g) CNV volumes quantified 7 days after combined laser injury and SRI of Aβ (P < 0.01, Mann-Whitney test. N = 6 (Aβ40−1), N = 7 (Aβ1−40))
The angiostimulatory activity of Alu RNA depends on inflammasomeAs inflammasome agonists may have pleiotropic effects aside from inflammasome activation, we next sought to determine whether this angiostimulatory effect was indeed dependent on inflammasome activity. To test this, we administered Alu RNA immediately following laser injury in mice lacking constituents of the inflammasome pathway, or in wild-type mice treated with pharmacologic inflammasome inhibitors.
The ATP receptor P2X7 is an upstream driver of inflammasome activation and is required for Alu RNA-induced RPE degeneration [23]. In P2rx7–/– mice, Alu RNA did not exacerbate laser-induced CNV (Fig. 2a). Nucleoside reverse transcriptase inhibitors (NRTIs) prevent P2X7-dependent inflammasome activation and RPE degeneration [14, 15, 24, 25]. Administering the NRTI zidovudine (AZT) into the vitreous of wild-type mice immediately following thermal laser burn and subretinal Alu RNA injection also abrogated the angiostimulatory response (Fig. 2b). In addition to targeting P2X7, NRTIs also inhibit reverse transcriptases. A 2-ethoxylated-modified derivative of AZT that does not inhibit reverse transcriptase but retains anti-inflammatory activities [25] also blunted Alu RNA-induced CNV (Fig. 2b), further supporting that the angiostimulatory activity of Alu RNA depends on P2X7.
Downstream of P2X7 activation, Alu RNA stimulates inflammasome assembly consisting of NLRP3, ASC, and the protease caspase-1 [2]. As in P2rx7–/– mice, CNV lesions in mice lacking NLRP3 (Nlrp3–/–) were also unaffected by Alu RNA (Fig. 2c). Conversely, mice lacking AIM2 (Aim2–/–), an alternative inflammasome receptor that does not mediate Alu RNA-induced RPE death (Fig S2), were susceptible to Alu RNA-induced exacerbation of CNV (Fig. 2d).
We next investigated the contribution of caspase-1, the inflammasome effector protease. Mice lacking both caspase-1 and the non-canonical inflammasome effector caspase-11 (Casp1–/–;Casp11–/–) are resistant to Alu RNA-induced RPE degeneration [2]. Similarly, Casp1–/–/Casp11–/– mice were resistant to the angiostimulatory activity of Alu RNA in CNV (Fig. 2e). In double knockout mice in which caspase-11 expression is rescued by a transgene (Casp1–/–; Casp11–/–;Tg+), Alu RNA treatment did not affect CNV (Fig. 2e), supporting that caspase-1 is essential for Alu RNA stimulated CNV. Furthermore, administration of Z-WEHD-FMK, a cell-permeable irreversible inhibitor of caspase-1, into the vitreous humor of wild-type mice also abrogated the angiostimulatory effect of Alu RNA on laser CNV (Fig. 2f). RPE degeneration by Alu RNA also depends on the activity of caspase-11 [26]. Mice lacking just caspase-11 (Casp11–/–) were partially protected against Alu RNA-induced CNV exacerbation (Fig. 2f), indicative of some contribution of non-canonical inflammasome activation to this process.
Inflammasome activation results in maturation of the effector cytokines IL-1β and IL-18, whose signal transduction requires the adaptor MyD88 [27]. Mice lacking MyD88 (Myd88–/–) are protected against Alu RNA-induced RPE degeneration [2]. In Myd88–/– mice, administration of Alu RNA did not affect CNV volume (Fig. 2g). Additionally, whereas Alu RNA induced excess CNV in eyes receiving a cell-permeable control inhibitor via intravitreous injection, administration of a MYD88 homodimerization peptide inhibitor [28] diminished the effect of Alu RNA on laser CNV (Fig. 2h).
Fig. 2
Intact NLRP3 inflammasome components are required for inflammasome agonism-dependent CNV exacerbation. (a) CNV volumes quantified 7 days after combined laser injury and SRI of Alu RNA in P2rx7–/– mice (P > 0.99, Mann-Whitney test. N = 7–8). (b) CNV volumes quantified 7 days after combined laser injury, SRI of Alu RNA, and intravitreous pretreatment with PBS (P < 0.01), AZT (P = 0.20), or K8 (P = 0.50) (two-way ANOVA. N = 6 per group). (c) Quantification of CNV volume 7 days post Alu RNA SRI in Nlrp3–/– mice (P = 0.412, Mann-Whitney test. N = 11 (PBS), N = 9 (Alu RNA)). (d) CNV volumes quantification 7 days after laser injury and SRI of Alu RNA in Aim2–/– mice (P < 0.01, Mann-Whitney test. N = 6 per group). (e) CNV volumes quantified after combined laser injury and SRI of Alu RNA in Casp1/11–/– (P = 0.25), Casp11–/– (P = 0.25), and Casp1/11–/– x Casp11Tg+ (P > 0.99) (two-way ANOVA, N ≥ 5 per group). (f) CNV volume quantification 7 days after combined laser injury, intravitreous administration of either control peptide Z-FA-FMK (P < 0.01) or caspase-1 inhibitor Z-WEHD-FMK (P = 0.98), and Alu RNA SRI (two-way ANOVA, N = 6 per group). (g) CNV volumes quantified after 7 days post-laser injury and Alu RNA SRI in Myd88–/– mice (P = 0.79, Mann-Whitney U test, N ≥ 5 per group). (h) CNV volumes quantified 7 days after combined laser injury, intravitreous administration of a peptide MyD88 inhibitor (P = 0.08) or control peptide (P < 0.01), and Alu RNA SRI (P = 0.08, two-way ANOVA, N ≥ 6 per group)
Collectively, these findings suggest that in the presence of an inflammasome agonist, inflammasome signaling amplifies pathological choroidal angiogenesis.
Inflammasome in myeloid cells is critical for Alu RNA-induced CNV exacerbationLaser CNV is a multicellular process involving multiple resident and recruited cells. Among these, circulating macrophages and neutrophils are recruited to CNV lesions and drive their growth [29, 30]. In Alu RNA-treated CNV lesions, immunofluorescent labeling revealed robust inflammasome activation via positive immunolabeling of the p20 subunit of caspase-1, which in part co-localized with CD11b+ macrophages (MΦ) (Fig. 3a). In addition, we observed substantial co-labeling of p20 and GFAP in Müller glia overlying the CNV lesion (Fig S3), though this p20/GFAP pattern was not as specific to the Alu RNA-treated CNV lesion as the p20/CD11b+. Therefore, we chose to assess whether inflammasome-dependent CNV expansion depends on inflammasome activation in myeloid cells, we generated myelomonocytic cell-specific caspase-1 knockout mice (LysM-Cre; Casp1loxP/loxP). We confirmed caspase-1 protein ablation by western blotting (Fig S4). Alu RNA-stimulated CNV was abrogated in LysM-Cre; Casp1loxP/loxP, but not in LysM-Cre-expressing control mice (LysM-Cre; Casp1+/+) (Fig. 3b), strongly suggesting inflammasome activation in myelomonocytic cells is responsible for Alu-RNA induced CNV aggravation.
Fig. 3
Inflammasome activation in myelomonocytic cells is crucial to laser CNV. (a) Representative immunofluorescence images of cross-section of mouse retinae treated with Alu RNA. Slides were stained with indicated antibodies. Scale bars: 50 μm. CC: choriocapillaris; RPE: retinal pigmented epithelium; SRS: subretinal space; ONL: outer nuclear layer (b) CNV volume quantification 7 days post laser injury and Alu RNA SRI in LysM-Cre (P = 0.03, N = 5) and Casp1f/f x LysM-Cre (P = 0.91, N = 6) mice (two-way ANOVA)
Inflammasome-dependent macrophage migration drives CNV exacerbationBased on the observation that inflammasome activation in myelomonocytic cells is required for inflammasome-induced CNV aggravation, we sought to assess whether inflammasome agonists and constituents affect macrophage recruitment in CNV. We quantified Mϕ migration in vivo by measuring the number of F4/80+ cells in Alu RNA-treated CNV lesions of WT and Nlrp3–/– mice. In the absence of an inflammasome agonist, CNV lesions from mice lacking Nlrp3 had similar F4/80+ immunolabeling cell count after laser injury compared to wild-type mice (Fig. 4a; representative images shown in Fig S5). Treatment with Alu RNA induced a greater number of CNV-associated F4/80+ cells in WT but not in Nlrp3–/– mice. Taken together, these findings suggest that inflammasome activation may mediate CNV exacerbation through the recruitment of immune cells.
To assess the contribution of inflammasome in macrophage recruitment, a Boyden chamber assay was used in which WT Mϕ were allowed to migrate towards a chemoattractant agent through a permeable support. As anticipated, a VEGF gradient stimulated robust MΦ migration (Fig. 4b). Neither DMSO nor Ac-YVAD-cmk, a cell-permeable caspase-1 inhibitor, impaired VEGF-induced chemotaxis, confirming that inflammasome inhibition did not affect the VEGF-induced chemotactic response (Fig. 4b). Next, we assessed whether inflammasome activation stimulates production of chemotactic signals. Conditioned media from Alu RNA-transfected wild-type MΦ stimulated chemotaxis to a similar degree as VEGF. However, conditioned media from Casp1–/–; Casp11–/– MΦ exhibited no detectable chemotactic activity following Alu RNA transfection (Fig. 4c). Similarly, conditioned media from WT MΦ pretreated with the caspase-1 inhibitor Ac-YVAD-cmk no longer exhibited Alu RNA-induced chemotactic activity (Fig. 4d). These findings indicate that inflammasome activation stimulates the production of soluble chemotactic factors.
Fig. 4
Inflammasome activation promotes chemotaxis in peripheral BMDM. (a) Macrophage number quantification after 3 days post laser injury and Alu RNA SRI in WT (P = 0.03, N = 7) and Nlrp3–/– (P = 0.85, N = 7, two-way ANOVA). (b) Relative migration of WT BMDM toward the following chemoattractants: VEGF, VEGF + DMSO, VEGF + Ac-YVAD-cmk (P < 0.001 compared to untreated cells, N = 4, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (c) Relative migration quantification of Alu RNA-transfected WT BMDM conditioned media (P < 0.01, N ≥ 8) and Alu RNA-transfected Casp1–/– BMDM (P = 0.97, N ≥ 4, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (d) Relative migration of WT BMDM with the following conditioned media as chemoattractant: Alu RNA-transfected WT BMDM (P < 0.01); untransfected WT BMDM pretreated with Ac-YVAD-cmk (P = 0.24); Ac-YVAD-cmk pretreated, Alu RNA-transfected WT BMDM (P = 0.43, N = 4, ordinary one-way ANOVA with Tukey’s multiple comparisons test)
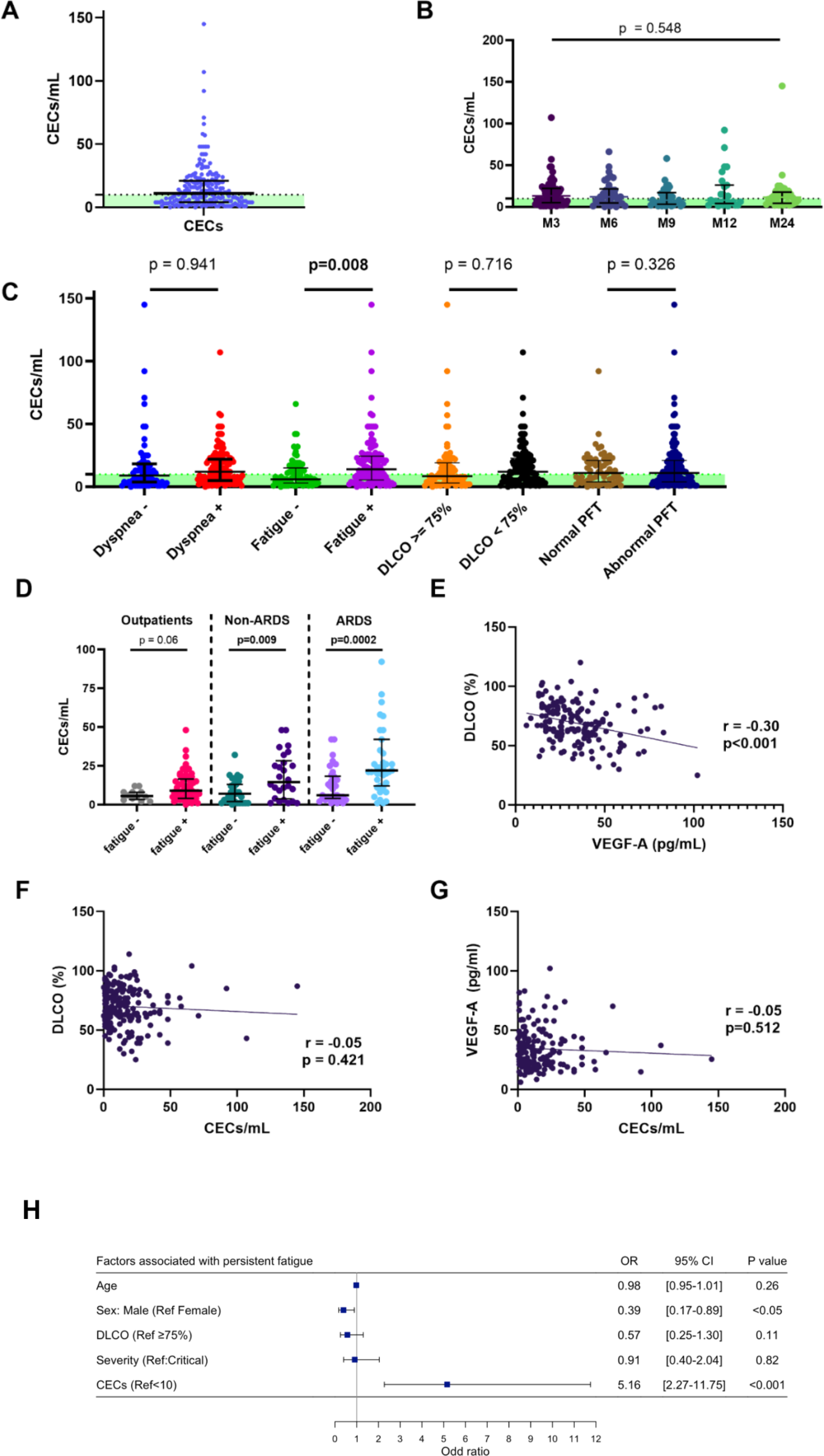
Inflammasome-induced macrophage migration is mediated by interleukin-1 beta (IL-1β)We sought to identify the inflammasome-dependent chemotactic factor responsible for macrophage migration. We focused on IL-1β as transfection of Alu RNA in WT MΦ induces its robust secretion [12] and it possesses chemotactic activity [31]. Conditioned media from Alu RNA-transfected WT BMDM incubated with an IL-1β neutralizing antibody (nAb) significantly inhibited WT BMDM chemotaxis (Fig. 5a). Consistent with a putative role in CNV, we detected robust Il1b mRNA expression that colocalized to Adgre1 mRNA (encoding the Mϕ marker F4/80) in CNV lesions of Alu RNA-treated eyes (Fig. 5b). Intravitreous administration of an IL-1β nAb reduced Mϕ accumulation (Fig. 5c) and day three CNV volume (Fig. 5d) in Alu RNA-treated eyes. We sought to determine whether combined administration of nAbs targeting Vegfa and IL-1β reduced CNV volumes in an additive manner. Intravitreous administration of nAbs against either Vegfa or IL-1β reduced day seven CNV volumes (Fig S7). Combined administration of low dose Vegfa nAb (1 ng) and IL-1β nAb reduced CNV volume to a greater extent than low dose Vegfa nAb alone; interestingly, combined administration of high dose Vegfa nAb (5 ng) and IL-1β nAb had no further reductive effect compared to high dose Vegfa nAb alone (Fig. S7). These findings suggest that while the effects of Vegfa and IL-1β inhibition appear to overlap, combining these two treatments may result in some increased therapeutic effect under specific conditions.
Mϕ inflammasome activation and IL-1β production could conceivably promote angiogenesis in two non-mutually exclusive ways. First, inflammasome agonists enhance Mϕ ingression (Fig. 4), which may be sufficient to drive increased angiogenesis. In addition, an inflammasome agonist could enhance the angiogenic potential of ingressed Mϕ. To test these concepts, an ex vivo choroidal sprouting assay was used as previously described [32, 33]. Three days after seeding choroid pieces from WT mice in growth factor-reduced Matrigel, an equal number of mock- or Alu RNA-transfected BMDM were added to each developing sprout and sprout size was quantified on day six. Consistent with previous reports [33], adding BMDM led to enhanced choroidal sprouting (Fig S6). However, Alu RNA-transfected BMDM did not further exacerbate sprout growth compared to mock-transfected BMDM (Fig S6). We interpret this finding to mean that inflammasome activation does not enhance the intrinsic angiogenic potential of BMDM, but rather exacerbates angiogenesis by increasing the extent of Mϕ ingression.
Fig. 5
IL-1β neutralization reduces Alu RNA-induced chemotaxis, macrophage accumulation, and laser CNV exacerbation. (a) Relative migration of Alu RNA-transfected WT BMDM conditioned media pretreated with either IgG (P < 0.01) or IL-1β neutralizing antibody (P < 0.01, N = 4, ordinary one-way ANOVA with Tukey’s multiple comparisons test). (b) Representative images of Alu RNA-treated laser CNV lesions hybridized with probes against Il1b and Adgre1. Scale bar: 50 μm. CC: choriocapillaris; RPE: retinal pigmented epithelium; SRS: subretinal space; ONL: outer nuclear layer; INL: inner nuclear layer (c) Macrophage number and (d) CNV volume quantification after 3 days post laser injury and Alu RNA subretinal injection with either 500 ng IgG1 or IL-1β neutralizing antibody (P < 0.001, N = 8–10)
留言 (0)