
記住我


We generated AdFITC-CAR T-cells to display a single-chain variable fragment (scFv) binding fluorescein tagged to fluorescein-labeled diabodies, acting as bridging adaptors, leading to cross-linking and conditional activation of the CAR T-cell against a target cell, Fig. 1A. We cloned the anti-fluorescein CAR gene, based on the E2 antibody clone, in a second-generation lentiviral vector incorporating CD8α hinge and transmembrane domain followed by the intracellular portions of 4-1BB and CD3ζ chains [23, 24]. The vector also drives expression of the RQR8 marker [33], which can be recognized both by the clinical-stage anti-CD20 antibody rituximab for CAR T-cell depletion in vivo, and by anti-CD34 antibodies, allowing for CAR T-cell identification and immunomagnetic positive selection Fig. 1B. CAR T-cells were transduced as previously described [23] and transduction rates were confirmed via flow cytometry, as shown for representative samples in Fig. 1C.
Fig. 1: Generation of AdFITC-CAR T-cells and fluorescein-labeled antibody constructs.
A Illustration of an AdFITC-CAR T-cell displaying anti-fluorescein scFv domain, targeting a generic tumor-associated antigen on a cell via a fluorescently labeled diabody adaptor as linking molecule. The surface co-expression of RQR8, which serves as a selection and depletion marker for AdFITC-CAR T-cells, is also depicted. B Schematic representation of second-generation CAR vector, presenting an anti-fluorescein scFv (clone E2). C Representative flow cytometry plots reflecting the transduction rate of healthy donor-derived T-cells are shown after staining with either AlexaFluor546-PEG2-FITC (AF546-FITC) in green or anti-CD34 antibody in red. Non-transduced T-cells served as control (black). D Plasmid map of the diabodies (Db) targeting CD33 and CD117 engineered to express a C-terminal cysteine. E Dbs were expressed and coupled with fluorescein-maleimide after a reduction step, as indicated in the protein conjugation scheme, yielding site-specifically labeled diabody (Db-FM). CD33 Db-FM (F) and CD117 Db-FM (G) were characterized by size exclusion chromatography (left), SDS-PAGE under non-reducing (NR) and reducing (R) conditions (middle; in addition, UV-light illuminated SDS-PAGE gels are also shown), and by mass spectrometry (right). M=Protein ladder indicating the kDa. Surface Plasmon Resonance analysis of CD33 Db-FM (H) and CD117 Db-FM (I) on a CM-5 chip coated with the extracellular domains of CD33 and CD117 His-tagged proteins, respectively. Dissociation constants KD are indicated. J Binding activity of the CD33 Db-FM and CD117 Db-FM was determined by dose titration on MOLM14-CD117highGFP+Luc+ cells and detected by means of an APC-conjugated anti-FITC antibody. MFI was normalized to account for the higher level of CD117 antigen expression. Mean ± SD from triplicates. Resulting KD,app are also indicated.
Due to the high phenotype similarities between healthy and malignant HSPCs [34] and aiming to use the AdFITC-CAR T-cell diabody approach as both time-limited leukemia eliminating and non-genotoxic conditioning prior to subsequent HSPC-transplantation, we hypothesized that CD33 and CD117 could be highly suitable target antigens [35, 36]. We validated their cell surface expression by flow cytometry on AML cell lines and patient AML blasts, as well as on healthy-donor HSPCs and T-cells, alongside a panel of selected AML antigens, including CD38, CD123, and CD371 [37,38,39], Fig. S1. Both, AML cell lines (Fig. S1A) as well as primary human AML cells (Fig. S1B) exhibited varying levels of CD33 and CD117 expression, with primary human AML cells showing a somewhat higher degree of positivity for CD117 than cell lines. HSPCs isolated from four healthy donors were analyzed upon lineage exclusion and divided in more immature Lin-CD34+CD38- and more mature Lin-CD34+CD38+ populations (Fig. S1C). While CD117 was similarly expressed in both populations, CD33 was up-regulated with HSPC maturation. Moreover, to gauge the risk of CAR T-cell fratricide, we analyzed the expression of these antigens on both stimulated and unstimulated T-cells (Fig. S1D). Apart from CD38, which was upregulated on stimulated T-cells, CD33 and CD117 expression was negative or comparatively low.
Based on these results and previously reported data [40,41,42], we confirmed and selected CD33 and CD117 for further AML targeting experiments and generated small antibody-based adaptors directed against CD33 and CD117. Considering that diabodies (Db) would be rapidly cleared from the body via the kidneys due to their molecular weight (~55 kDa), allowing for rapid control over AdFITC-CAR T-cell on-off activity on-demand, we cloned Db with C-terminal reactive cysteine residues [43, 44] (Fig. 1D). The respective antibody sequences are shown in Fig. S2. The diabodies were subsequently conjugated by site-specific labeling of the engineered cysteines with fluorescein-maleimide, leading to Db-FM conjugates, Fig. 1E. Db-FM adaptors were characterized by size-exclusion chromatography, SDS-PAGE with fluorescence detection, and mass spectrometry, confirming that constructs could be produced and fluorescently labeled, maintaining high levels of purity, Fig. 1F, G. Binding of the adaptors was confirmed by BIAcore analyses on chips coated with cognate antigens with dissociation constants in the low nanomolar range (Fig. 1H, I) and by staining MOLM14-CD117highGFP+Luc+ AML cells, positive for CD33 and CD117, exhibiting semi-saturation in the low nanomolar range (Fig. 1J). From this data, we conclude that this approach represents a viable strategy to generate AdFITC-CAR T-cells as well as highly homogeneous fluorescein-conjugated Db products with complete retention of target binding properties.
AdFITC-CAR T-cells mediate tumor cell lysis dependent on the density of both target antigens and target-bound fluoresceinated adaptorsWe hypothesized that similarly to conventional direct CAR T-cells, also AdFITC-CAR T-cells would exhibit cytotoxic activity dependent on target antigen density [7, 23]. In our work, this effect might depend on the levels of target-antigen (CD33 and CD117) expression on the tumor cell surface, as well as on the density of the CAR T-cell ligand, fluorescein, decorating the tumor cell surface. To confirm expression levels of the target antigens on the three AML cell lines Kasumi-1, endogenously expressing CD33 and CD117, HL-60, and MOLM14, both expressing CD33 and transduced to display the extracellular domain of CD117 and sorted based on its expression level [23], we stained them with CD33 or CD117 Db-FM and detected the antigen-bound Db-FM with an APC-conjugated anti-FITC antibody (Fig. 2A, C, E). Next, we co-cultured AdFITC-CAR T-cells and titrations of both Db-FM in short-term in vitro experiments with Kasumi-1, HL-60, and MOLM14 cells, expressing various levels of CD117 target antigen (Fig. 2B, D, F). The resulting target cell lysis increased over time, depending on the concentration of adaptors, with EC50 in the sub-nanomolar range. As expected, CD117 antigen density affected AdFITC-CAR T-cell efficiency. Although the lysis of HL-60 CD117high showed a 8-fold lower EC50 than HL-60 CD117mid and 30-fold lower than HL-60 CD117low, a more moderate difference was observed in MOLM14 clones expressing various levels of CD117, where EC50 resulted to be 100-fold lower than HL-60 cells, regardless of the Db-FM. In line with this, the release of pro-inflammatory cytokines IL-2 and IFN-γ was positively correlated with the lytic activity of the CAR T-cells (Fig. 2G, H). Collectively, these data demonstrated that AML cell lysis and AdFITC-CAR T-cell activation were positively correlated with the amount of fluorescein-labeled Db adaptors and with antigen density on target cells.
Fig. 2: Db-FM adaptors elicit AdFITC-CAR T-cell cytotoxicity against various human AML cell lines in a concentration, time, and antigen density-dependent manner.
A Representative histograms showing Kasumi-1 GFP+ cells stained with 10 nM CD33 Db-FM (left) and CD117 Db-FM (right), detected by APC-conjugated anti-FITC antibodies. MFI is indicated. B Kasumi-1 and AdFITC-CAR T-cells were co-cultured at an effector-to-target ratio E:T-1:1, tumor cell lysis was assessed via flow cytometry after 24, 48, and 72 h of co-culture. Percentage specific lysis of Kasumi-1 cells is shown relative to Db-FM concentration, represented as mean ± SD from three healthy-donor-derived AdFITC-CAR T-cell donors, each plated in duplicate wells. C Representative histograms showing HL-60 GFP+ cells transduced to express CD117 at high, medium, and low levels, labeled with 10 nM CD33 Db-FM (top) and CD117 Db-FM (bottom) as described in (A). D Percentage specific lysis of HL-60 cells was assessed over time after co-culture with AdFITC-CAR T-cells at E:T-1:1 and increasing levels of Db-FM. Mean ± SD from three healthy-donor AdFITC-CAR T-cell donors, each plated in duplicates. E Representative histograms showing MOLM14 GFP+ cells transduced to display various levels of CD117, stained and analyzed as described in (C). F MOLM14 cell lysis mediated by AdFITC-CAR T-cells at E:T-1:1 in culture with Db-FM. Percentage specific lysis of MOLM14 cells as a function of Db-FM concentration is represented as mean ± SD from three healthy-donor-derived AdFITC-CAR T-cell donors plated in duplicate wells. Quantification of IL-2 (G) and IFN-γ (H) in the supernatant from the same co-culture experiment indicated in (F) at 24 h.
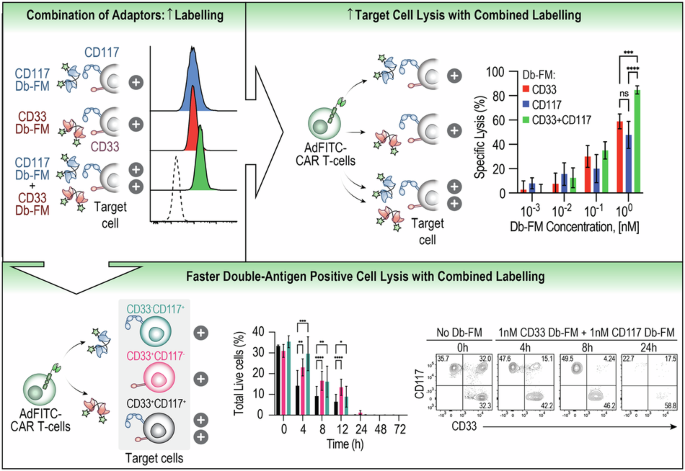
Combinatorial use of Db-FM adaptors increases biocidal activity of AdFITC-CAR T-cells against AML cell lines expressing the target antigensThe observation that AdFITC-CAR T-cell biocidal activity was dependent on the adaptor concentration in our and previous results [7, 23] suggested that local increase of CAR-target (fluorescein) density, such as by simultaneous use of multiple tagged adaptors, could enhance the lytic efficacy. To test this hypothesis, we stained a cell line positive for both antigens, MOLM14-GFP-CD117mid, with saturating concentrations of CD117 and CD33 Db-FM as individual agents or in combination (Fig. 3A). As expected, we detected more than two-fold increased fluorescence upon combined staining (MFI = 924, 942 and 1918, respectively; Fig. 3B). Next, we interrogated whether increased fluorescein staining achieved by combinatorial use of multiple adaptors would also correlate to higher cytotoxic activity. Indeed, we observed enhanced tumor cell lysis when CD117 and CD33 Db-FM were added compared to equimolar concentrations of single agents alone (Fig. 3C). To further investigate the basis for increased lysis by dual targeting, we performed live cell imaging of effector-target cell interactions in microwells (Fig. 3D). The addition of both CD117 and CD33 Db-FM to the co-culture of MOLM14 CD117mid and AdFITC-CAR T-cells resulted in significant faster tumor cell lysis (measured by influx of PI) as shown in Fig. 3E, which correlated with a more rapid, durable engagement time of CAR T-cells with target cells (Fig. S3A–E). Interestingly, no significant difference was observed in the time from initial durable engagement to lysis (Fig. S3D–E). Given the theoretical antigen-downregulation AML blasts could undergo during single-adaptor treatment, leading to escape of antigen-negative clones, we assessed the effect of the combination of adaptors on a heterogeneous tumor population. To test this, we titrated single Db-FM adaptors or equimolar concentrations of both adaptors on a pool of three transduced MOLM14 cell lines, engineered to express either CD117, or CD33, or both antigens (i.e. MOLM14-CD117highCD33KO, MOLM14-CD117negCD33high and MOLM14-CD117highCD33high) and recorded lytic activity mediated by AdFITC-CAR T-cells (Fig. 3F). The combinatorial use of 1 nM CD33 Db-FM and 1 nM CD117 Db-FM resulted in a significant increase in overall tumor cell lysis (Fig. 3G, H). Notably, despite the use of different cell lines in combination within the same culture wells, the lytic activity was primarily directed against cells expressing the cognate antigen of the specific adaptor utilized. Considering the correlation between enhanced cytotoxicity and increased surface-bound fluorescein, we hypothesized that combinatorial Db-FM treatment would lead to increased fluorescein staining of double-antigen-positive cells and enhanced lysis even in the presence of bystander single-antigen expressing cells. We thus co-cultured AdFITC-CAR T-cells in combination with CD33 and CD117 Db-FM against the same equal numbers of MOLM14 cells used in Fig. 3G, H and analyzed combined population lysis (Fig. 3I) alongside the sub-populations (Fig. 3J, K) over 72 h. At early time points after seeding (4, 8, 12 h), we observed significantly increased ablation of tumor cells expressing both CD33 and CD117 compared to only single-antigen-positive cells. At later time points, this effect was not detectable anymore when overall tumor cell lysis reached >90%.
Fig. 3: Combination of diabody adaptors enhances cytotoxicity against AML cell lines compared to single adaptors.
A Experimental setup to evaluate adaptor-mediated AdFITC-CAR T-cell activity against MOLM14 cells, double positive for the targeted antigens CD33 and CD117. B MOLM14 GFP-CD117midCD33+ cells were stained with 10 nM of either CD33 Db-FM, CD117 Db-FM, or both Db-FM (10 nM each). Unstained cells served as negative control. C Equal ratio of MOLM14-CD117midCD33+ cells and AdFITC-CAR T-cells were cultured with increasing concentrations of CD33 and CD117 Db-FM, alone or in combination. Percentage specific lysis after 24 h is shown. Statistical analysis at the indicated EC50 was performed with one-way ANOVA, ***p < 0.001. D, E Time-lapse imaging to assess MOLM14-CD117midCD33+ cell lysis by AdFITC-CAR T-cells in combination with CD33 and/or CD117 Db-FM. Time to PI influx was significantly shorter with the combinatorial adaptor approach when compared to single adaptor targeting (results from two healthy donors, statistical analysis conducted using one-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). F Experimental setup to evaluate adaptor-mediated AdFITC-CAR T-cell activity against MOLM14 cells, being either single or double positive for the targeted antigens. G, H MOLM14-CD117negCD33+, MOLM14-CD117highCD33+, and MOLM14-CD117highCD33KO cells were mixed at equal ratios and co-cultured with AdFITC-CAR T-cells at an E:T = 1:1. CD33 and CD117 Db-FM were added at 1 nM as single agents or in combination. G Percentage specific lysis of the target cells was measured at indicated time points. Statistical analysis was conducted using two-way ANOVA; ***p < 0.001; ****p < 0.0001. H Representative flow cytometry plots showing residual MOLM14 subpopulations at 24 h. I–K MOLM14-CD117negCD33+, MOLM14-CD117highCD33+, and MOLM14-CD117highCD33KO cells were mixed at equal ratios and co-cultured with healthy donor-derived AdFITC-CAR T-cells were mixed as in (G, H). The combination of CD33 and CD117 Db-FM was added at 1 nM, and target-cell lysis was measured at indicated time points. Percentage specific lysis of the overall target cells (I) and subpopulations (J) at the indicated time points. Statistical analysis was conducted using two-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. K Representative flow cytometry plots showing residual MOLM14 subpopulations at 0, 4, 8, and 24 h. A–K Unless otherwise indicated, data are presented as mean ± SD from three healthy donor-derived AdFITC-CAR T-cells, each plated in duplicate wells.
These findings provide compelling evidence that a combination of adaptors targeting multiple tumor antigens contributes to enhanced AdFITC-CAR T-cell efficacy by increasing the fluorescein decoration of target cells, thereby overcoming limitations posed by the density of single antigen expression and possibly extending the targeting to heterogeneous antigen expressing tumors.
Combination of Db-FM adaptors enhances AdFITC-CAR T-cells lysis of primary human AML blastsGiven that the target antigens CD33 and CD117 are expressed with intra- and inter-patient heterogeneity on primary human AML blasts, we next investigated the efficacy of Db-FM and AdFITC-CAR T-cells against AML patient blasts in vitro. At thawing and after 24 h in culture with AdFITC-CAR T-cells, target cells exhibited a varied expression pattern of CD33 and CD117 (Fig. 4A). When effector and target cells were cultured together in the presence of defined concentrations of Db-FM, AdFITC-CAR T-cells were able to eliminate antigen-positive AML subpopulations, while sparing antigen-negative ones. The addition of both Db-FM, resulted in the lysis of the majority of blast cells, leading to a significantly improved cytotoxicity compared to equivalent concentrations of single Db-FM (Fig. 4A–C). This further supports the concept that combinatorial use of adaptors can substantially enhance the cytolytic efficacy of AdFITC-CAR T-cells, also against primary human AML blasts.
Fig. 4: Combinatorial targeting of primary patient AML blasts is more efficient than single-adaptor targeting.
A Surface expression of target antigens (CD33 and CD117) on CD45dimCD3- cells from three AML patient samples. Representative flow cytometry analysis upon thawing, before and after the addition of healthy donor AdFITC-CAR T-cells at E:T = 1:1 ratio and after 24 h incubation in the presence of the indicated concentrations of diabody adaptors is shown. Percentage specific lysis of CD45dimCD3- AML blast cells after 24 h (B) and 48 h (C) in culture with Db-FM targeting CD33 and CD117, alone or in combination. Data from 2 independent experiments with AML cells from three patients and AdFITC-CAR T-cells derived from 3 different healthy donors plated in duplicates (mean ± SD). Statistical analysis was conducted using two-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
In vitro and in vivo activation and cytokine release of AdFITC-CAR T-cells in absence of target-antigen expressing cellsConsidering the observation that ligand-mediated dimerization can trigger CAR signaling even without the presence of antigen-positive cells [45, 46], we evaluated the induced AdFITC-CAR T-cell activation from crosslinking with bivalent Db-FM in absence of target-antigen expressing cells in vitro and in vivo. We incubated CAR T- and control T-cells, isolated from the same healthy donors and previously expanded in IL-2, in vitro for 72 h with or without addition of 10 nM CD117 Db-FM (Fig. S4A, B). We monitored their activation status at 24, 48 and 72 h by flow cytometry and compared to conditions where MOLM14-CD117high target cells were added at an E:T = 1:1 to the co-culture (Fig. S4C, D). An upregulation of CD69+CD25+ was observed for RQR8+ AdFITC-CAR T-cells in culture with CD117 Db-FM with a progressive polarization towards CD69-CD25+ over time, especially visible in culture with target cells. The activation was accompanied by cellular clustering (Fig. S5A) and proliferation (Fig. S5B). In stark contrast however, significant levels of both IL-2 and IFN-γ were only released at high levels (10- and 280-fold increase, respectively) in presence of CD117-expressing MOLM14 cells (Fig. S5C). Importantly, neither Db-FM cultured with T-cells nor AdFITC-CAR T-cells without Db-FM resulted in T-cell activation, cytokine release and tumor cell lysis (Fig. S6A), indicating that biocidal activity was conditionally and selectively triggered by the concomitant presence of AdFITC-CAR T-cells and Db-FM.
We further evaluated AdFITC-CAR T-cell activation in vivo following multiple injections of CD117 Db-FM, which is not cross-reactive with murine CD117 [28]. NSG mice were injected i.v. with 107 AdFITC-CAR T-cells followed by i.p administration of 25 μg Db-FM every 12 h, for a total of 4 doses over 48 h (Fig. S7A). Fluorescein was detectable on all AdFITC-CAR T-cells harvested from peripheral blood (PB), BM and spleen, and positively correlated with RQR8 expression (Fig. S7B). Importantly, the presence of Db-FM in vivo did not result in AdFITC-CAR T-cell activation nor T-cell expansion and did not lead to elevated levels of pro-inflammatory cytokines (Fig. S7C–F). Thus, although the presence of adaptors in absence of the target-antigen led to phenotypic AdFITC-CAR T-cell activation without cytokine production in vitro, this activation was not detectable in vivo when using biologically active Db-FM concentrations, suggesting that adaptor-mediated activation of AdFITC-CAR T-cells in the absence of target antigen likely will not pose a relevant toxicity in vivo.
Db-FM adaptors exhibit short in vivo serum half-lives while maintaining longer on-tumor residence timeTo study the in vivo half-life of Db-FM adaptors, we administered 25 μg of CD117 Db-FM either intravenously (i.v.) or intraperitoneally (i.p.) to NSG mice and collected blood 1 min post-injection and terminally at the indicated time points (Fig. 5A). Serum half-life accounted for approximately 30 minutes upon i.v. injection and 40 minutes for i.p. injection, with possibly slightly longer exposure to Db-FM following i.p. injections, as indicated by the values of area under the plasma concentration-time curve (AUC) in Fig. 5B. Before studying the residence time of Db-FM:tumor complex in vivo, we assessed the growth kinetics of MOLM14 cells in NSG mice at two different doses (1 and 2.5 × 105) by correlating bioluminescence flux with terminal flow cytometry analysis and immunohistochemistry staining (Fig. S8A–E). Remarkably, flow analysis of BM underestimated MOLM14 engraftment levels when compared to histology analysis (Fig. S8E). Based on our data, injection of 1 × 105 consistently resulted in detectable bioluminescence signals by day 7, with formation of tumor cell infiltrates in the bone marrow (BM), confirmed by HE and CD117 immunohistochemistry staining. To determine the in vivo residence time of the Db-FM adaptor on tumor cells, we injected 50 μg of CD117 Db-FM in NSG mice, engrafted ten days prior with 1 × 105 MOLM14-CD117high GFP+Luc+ cells (Fig. 5C). Db-FM was detectable on MOLM14 cells in decreasing intensity for up to 12 h after i.v. or i.p. injection by flow cytometry (Fig. 5D). This decrease of in vivo labeling over time is likely due to the concomitant effects of the Db-FM off-rate and fast Db-FM clearance as well as the exponential growth of MOLM14 cells in vivo and therefore dilution of surface-bound Db-FM (Fig. S8C, D).
Fig. 5: In vivo pharmacokinetic (PK) and pharmacodynamic (PD) properties of Db-FM adaptors.
A Schematic representation of PK experimental set-up in mice not carrying target cells. NSG mice received a single i.v. or i.p. injection of 25 μg CD117 Db-FM (n = 2 mice per time point per administration route). Blood was collected from all mice at 1 min post-injection and terminally at the indicated time points. B Db-FM concentration in serum was determined by ELISA (mean ± SD of technical duplicates per mouse). Calculated t1/2 and AUC are indicated. C Experimental set-up to study on-tumor residence time of CD117 Db-FM on MOLM14-CD117high GFP+Luc+ cells in xenografted mice. NSG mice were sublethally irradiated and injected i.v. with 105 MOLM14 cells. On day 10, 50 μg of CD117 Db-FM were injected i.v. or i.p. (n = 1 mouse per time point per administration route). D At the indicated time points, live MOLM14 cells (hCD45+GFP+) isolated from BM were analyzed by flow cytometry (representative dot plot on the left), and target-bound CD117 Db-FM was detected by APC-conjugated anti-FITC antibody staining (histograms on the right). MOLM14 cells from mice engrafted but not subsequently injected with Db-FM served as negative (−) and positive controls after ex vivo staining with 10 nM CD117 Db-FM (+). E Sublethally irradiated NSG mice were injected i.v. with 105 MOLM14-CD117highGFP+Luc+ cells. On day 10, 50 μg of CD117 or CD33 Db-FM were injected i.v. and femora were collected after 1 and 6 h to determine the clearance of Db-FM from the tumor cell surface by flow cytometry (n = 1 mouse per time point per Db-FM). A mouse engrafted with MOLM14 but not injected with adaptors served as negative control. F, G After ex vivo staining of cells isolated from the control mouse with titration of Db-FM, the surface labeling levels obtained from known Db-FM concentrations were used to interpolate the concentrations of Db-FM on tumor cells retrieved from mice injected with Db-FM. MFI resulting from target-bound Db-FM was detected by APC-conjugated anti-FITC antibody. F Interpolated on-tumor CD33 Db-FM concentration was estimated at 0.99 nM after 1 h injection and 0.22 nM after 6 h (each indicated in red). G Interpolated on-tumor CD117 Db-FM concentration was determined to be 4.37 nM after 1 h injection and 1.37 nM after 6 h (each indicated in blue).
We next evaluated the in vivo tumor-residence time of both adaptors by injecting tumor-bearing mice with CD33 or CD117 Db-FM (Fig. 5E). The intensity of MOLM14 cells retrieved from mice injected with Db-FM 1 and 6 h prior to terminal analysis, was compared to the staining from ex vivo titrations of CD33 or CD117 Db-FM (Fig. 5F, G). Assuming an average plasma volume of 49 μl/g [47] and an immediate distribution in the blood compartment, the theoretical initial concentration of Db-FM in serum accounted on average for 744 nM. Based on the interpolated concentration of tumor-bound Db-FM (Fig. 5F, G), CD117 Db-FM displayed higher MFIs (consistent with the antigen expression levels), with on-tumor concentrations starting at 4.4 nM 1 h after injection, with a 3-fold decrease observed between 1 and 6 h post-injection. Conversely, CD33 Db-FM presented a relatively lower density on the tumor cell surface (1 nM) and experienced a 4.5-fold reduction in surface labeling over the same time, possibly explained by receptor-mediated internalization of CD33 Db-FM [48].
Based on the determined on-tumor residence time of Db-FM, we designed a dosing regimen with i.p. Db-FM injections scheduled at 12 h intervals, aiming to maintain sufficient fluorescein density on MOLM14 cell surface and counteract the rapid clearance from the tumor cell surface during the in vivo therapy phase.
AdFITC-CAR T-cells in combination with CD33 or CD117 Db-FM deplete MOLM14 cells as efficiently as direct antigen-targeting CAR T-cells in vivoWe next tested AdFITC-CAR T-cell cytotoxic activity in vivo. Following confirmation of MOLM14-CD117highGFP+Luc+ engraftment at day 7 by bioluminescence imaging of NSG mice, we administered i.v. either 107 direct anti-CD33 or anti-CD117 CAR T-cells, or 107 AdFITC-CAR T-cells followed by 25 μg CD33 or CD117 Db-FM i.p. every 12 h, respectively (Fig. 6A). At day 19, mice were sacrificed as untreated control mice met termination criteria. Bioluminescence imaging showed inhibition of tumor growth in mice treated with direct CAR T-cells and AdFITC-CAR T-cells in combination with the respective adaptors (Fig. 6B, C, E, F). At terminal analysis, we observed successful eradication of MOLM14 cells in the BM of treated mice by flow cytometry and reconstitution of visually relatively normal marrow by histology analysis (Fig. 6D, G–I). Some mice exhibited low-level extra-medullary engraftment of MOLM14 cells, as detected by bioluminescence imaging, in both the direct and adaptor-mediated CAR T-cell experimental groups. Importantly, deviation from the 12 h administration schedule, such as administering antibody every 48 h, abolished the therapeutic effect (Fig. S9A–C), indicating that maintaining continuous and adequate coverage of adaptors on the surface of tumor cells is crucial for therapeutic success. These data collectively underscore the comparable efficacy of AdFITC-CAR T-cells in combination with their respective adaptors and direct CAR T-cells in xenogeneic therapy studies. Moreover, considering the tumor engraftment in presence of AdFITC-CAR T-cells but absence of adaptors, this suggests that the system would allow for off-control of AdFITC-CAR T-cell function in vivo.
Fig. 6: In vivo therapeutic efficiency of direct CAR T-cells and AdFITC-CAR T-cells in combination with CD33 or CD117 Db-FM adaptors against MOLM14-CD117highAML cells.
A Schematic outline of the experimental setup. Sub-lethally irradiated mice were engrafted with 105 MOLM14-CD117highGFP+Luc+ cells. Seven days later, engraftment was confirmed by bioluminescence analysis (BLI), and mice were injected with either 107 direct CAR T-cells targeting CD33 or CD117 (positive controls) or AdFITC-CAR T-cells (n = 3/4 mice per group). Mice from the latter group subsequently received 25 μg CD33 Db-FM or CD117 Db-FM (i.p. every 12 h) or no adaptors (negative controls). B Bioluminescence analysis of MOLM14 cell engraftment at days 7, 14, and 19 in mice treated with PBS, AdFITC-CAR T-cells in the absence or presence of adaptor, or direct CAR T-cells (CD33-CAR). C Quantification of the bioluminescence flux of whole-body imaging of mice in (B). D Absolute counts of hCD45+GFP+ MOLM14 cells (mean ± SD) in the BM of a single femur per mouse at terminal analysis. Statistical analysis was conducted using two-way ANOVA; *p < 0.05, **p < 0.01. E Bioluminescence analysis at indicated time points of mice treated with PBS, AdFITC-CAR T-cells with or without adaptor, or direct CAR T-cells (CD117-CAR). F Corresponding quantification of the bioluminescence flux of mice showed in (E). G Absolute hCD45+GFP+ MOLM14 cell count (mean ± SD) in the BM of a single femur per mouse at the end of the study, statistical analysis performed as indicated in (D). B–G Data shown from two independent experiments. H Representative flow cytometry plots of live cells from BM single-cell suspensions receiving the above-indicated treatment. I HE staining of the contralateral femora from the mice depicted in (H), as well as from healthy age-matched control mice (20x magnification, solid line indicates 100 μm).
We next evaluated the potency of AdFITC-CAR T-cells by limiting their dose in vivo, starting from 107 and decreasing fivefold down to 0.2 × 107. A reduction in CAR T-cell numbers resulted in a reduced therapeutic response, as assessed by bioluminescence imaging, with a more pronounced effect in the context of adaptor-CAR T-cell approaches compared to direct CAR T-cells (Fig. S10A–D). These findings highlight the importance of establishing a sufficient effector-to-target ratio at start of therapy. Considering the estimated doubling time ranging here from 0.6 and 0.9 days, as determined from tumor growth kinetics based on bioluminescence signal and flow cytometry (Fig. S8C, D), it can be estimated that at least 9 × 106 MOLM14 target cells would be present in mice at the start of the therapy, leading to an E:T of about 1:1 when 107 CAR T-cells are transferred. In sum, these findings underline that the effectiveness of AdFITC-CAR T-cells in our exemplary model system is, as expected, dependent on both the dosage of CAR T-cells and a regimen enabling their sustained activation in vivo.
Combinatorial use of adaptors leads to most efficient AdFITC-CAR T-cell mediated AML cell line elimination in a therapeutic setting in vivoBuilding on these findings, we sought to evaluate the therapeutic efficacy of combinations of adaptors in vivo against an AML cell line. First, we investigated whether simultaneous administration of both adaptors would also result in increased fluorescein intensity on tumor cells isolated from the BM at terminal analysis, similarly to what we already observed in vitro (Fig. 3A). NSG mice were engrafted with MOLM14-CD117highGFP+Luc+ cells, and 10 days later, injected i.v. with 50 μg of CD33 and CD117 Db-FM alone or in combination (Fig. 7A). Indeed, concurrent administration of both adaptors resulted in the highest MFI (Fig. 7B), recapitulating the delta in intensity observed after ex vivo staining of cells isolated from a mouse not receiving Db-FM (Fig. 7C). Next, we treated tumor-bearing mice with AdFITC-CAR T-cells and either 12.5 μg CD117 or CD33 Db-FM as monotherapies or a combination of both adaptors for three weeks starting from seven days after MOLM14-CD117highGFP+Luc+ injection (Fig. 7D). At day 28, mice treated exclusively with CD33 and CD117 Db-FM experienced the occurrence of extramedullary manifestations as myeloid sarcomas. Conversely, in mice treated with a combination of Db-FM, tumor growth was effectively inhibited as indicated by a lower overall bioluminescence signal (Fig. 7E, F). Flow cytometry analysis at the end of the experiment indicated that AdFITC-CAR T-cells with adaptors (alone or in combination) reduced the tumor engraftment in BM and blood (Fig. 7G) accompanied by expansion and persistence of AdFITC-CAR T-cells after infusion (Fig. 7H). Together, these data demonstrate that combinatorial targeting of antigens with adaptor-based CAR T-cell strategies leads to more efficient AML cell elimination in vivo.
Fig. 7: Combinatorial use of adaptors improved in vivo therapeutic efficacy of AdFITC-CAR T-cells against MOLM14-CD117high AML cells.
留言 (0)