Sources of database informationData of antitumor pCms
Information on antitumor pCms is retrieved from China’s Catalogue of Drugs for Basic National Medical Insurance/Employment Injury Insurance/Birth Insurance (2022) (NRDL). The antitumor pCms included in the NRDL are widely applied in clinical treatment. These medicines have undergone multiple clinical trials, demonstrating significant therapeutic effects, minimal side effects, and ensuring clinical efficacy and medication safety for patients. Moreover, inclusion in the NRDL also indicates a pricing advantage, which benefits patients greatly.
Data of Chinese materia medica
Information on Chinese materia medica is sourced from the Pharmacopoeia of the People’s Republic of China 2020 (Ch.P), TCMSP, ETCM, and TCM Miner. The Ch.P represents the most authoritative codex for traditional Chinese medicine data as an essential component of China’s national drug standards, containing 616 commonly used Chinese herbal medicines. The TCM Miner is an information retrieval platform developed by the Institute of Traditional Chinese Medicine Information of China Academy of Chinese Medical Sciences, collecting Chinese herbal medicine’s basic information, local standards, and national standards.
Data of chemical components
Information on the chemical components of Chinese herbal medicine primarily comes from databases such as PubChem, TCMSP, TCM-ID, and HERB. PubChem, developed by the United States National Institutes of Health, is an open-source database that includes information on small molecule compounds’ physicochemical properties, biological activities, toxicities, and patents [14]. A comprehensive search is conducted within the DrugBank database using the obtained component entity information, acquiring additional component entities information, including molecular formulae, SMILES codes, and indications [15].
Data of targets
Primary data sources for target entities include databases such as TCMSP, SymMap, SwissTargetPrediction, NCBI Gene, HERB, and others. SwissTargetPrediction predicts molecular targets based on the SMILES code or structural formulas of small molecules [16]. NCBI Gene, a gene data platform developed by the United States National Center for Biotechnology Information, includes extensive gene attribute and entity information [17].
Disease data
Disease entity data mainly originates from DisGetNET and GeneCard databases. DisGetNET consolidates human disease-related gene information from various disease databases, including OMIM, HPO, UMLS, and MeSH. GeneCard, a free, comprehensive database available to research institutions, has integrated information from 193 public gene-associated databases as of December 1, 2023, and offers a wealth of transcriptome, genome, and proteome data [18]. In this study, diseases corresponding to pCms were determined based on their leaflets and relevant literature concerning their modern clinical applications. Due to possible inconsistencies in gene or protein IDs from different source databases, manual examination and conversion into official gene IDs and protein Uniprot IDs were performed.
Entity relationship and network construction
Integrating entity data from various databases results in three types of entity relationships: pCms–Chinese herbal medicines, Chinese herbal medicine–components, and components–targets. The network module within the vis.js dynamic browser-based database was employed to construct user-friendly networks based on the entity relationships among pCms, Chinese herbal medicines, components, and targets. These networks were then visualized on the database’s corresponding pages.
Construction of antitumor pCms–component–target network
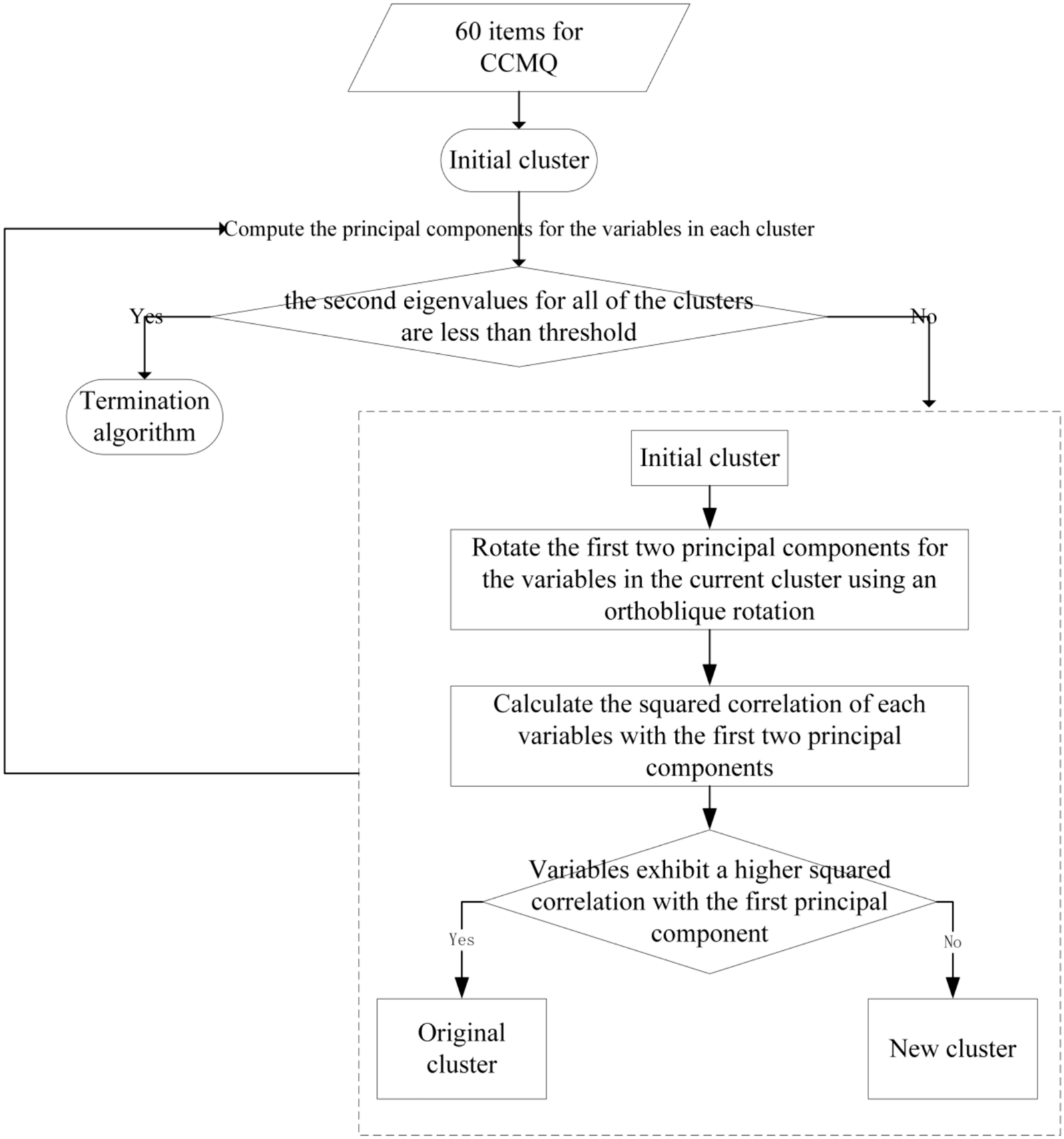
By using bibliometric approach, preliminary research has identified several antitumor pCms which are widely adopted in GC treatment (Additional file 1). In this study, these antitumor pCms’ names were searched within the databases to obtain their formulation, followed by searching for corresponding chemical component and target data. The target information for GC was acquired by searching the antitumor pCm database using “Gastric Cancer” as the keyword. Utilizing information on pCms, Chinese herbal medicines, components, and targets, the network files and type files were prepared and imported into Cytoscape software for network visualization. This process resulted in the construction of the antitumor pCm–component–target network. Network topology analysis was then conducted, and Degree values were derived for subsequent analyses.
Construction of PPI network for GC treatment with antitumor pCms
Intersection targets between antitumor pCm targets and GC targets were determined and visualized using a Venn diagram. The intersection targets were analyzed in the STRING database (https://cn.string-db.org/), setting the minimum required interaction score to 0.4, displaying only connected network nodes, and obtaining the.tsv file of the PPI network. This file was then imported into Cytoscape software for PPI network visualization and subsequent network topology analysis, with key targets identified based on Betweenness, Closeness, and Degree values.
Molecular docking
Initially steps involved obtaining chemical component nodes with a Degree > 100 from the antitumor pCm–component–target network, which were considered as the network’s core components. Key targets were then screened based on the topological parameters of the PPI network. Core components and key targets were assumed to be ligands and receptors, respectively, and used for molecular docking. Molecular docking calculations were performed using Autodock Vina 1.1.2. The binding free energies between core chemical components and key targets were ranked in ascending order. A molecular docking heatmap was generated using the pheatmap package in R, and core chemical components with minimum binding energy levels to bind to the key targets were selected. Docking results were visualized using PyMOL 2.3.2 software.
Cell lines, cell culture, and experimental drugs
The human GC cell line HGC-27, acquired from Wuhan Procell Life Science & Technology Co., Ltd., was verified by STR identification. Cells were routinely cultured in RPMI-1640 complete medium (Procell, China) at 37 °C, 5% CO2, and saturated humidity. Norcantharidin (Purity ≥ 98%) was procured from Shanghai YuanYe Biotechnology Co., Ltd.
CCK-8 assay
Cells were digested with a 0.25% trypsin solution (containing EDTA) and seeded in 96-well plates at a density of 5.0 × 104 cells/mL, with 100 μL per well and 6 replicates per group for 24 h. For each well, complete medium containing norcantharidin at concentrations of 1.25 μM, 2.5 μM, 5 μM, 10 μM, 20 μM, 40 μM, 80 μM, and 160 μM was added and cultured for an additional 24, 48, and 72 h. At the end of the culture period, 100 μL of CCK-8 diluent (culture medium: CCK-8 reagent = 9:1) was added to each well, incubated in a 37 °C incubator for over 30 min, and the OD value at 450 nm was determined using a microplate reader. A cell viability curve was then plotted.
Colony formation assay
Cells were digested with a 0.25% trypsin solution (containing EDTA) and seeded in 6-well plates at a density of 5.0 × 102 cells/mL, with 1 mL per well and a 24 h culture period. The control group was provided with complete medium, while treatment groups received complete medium containing norcantharidin at concentrations of 15 μM, 30 μM, and 60 μM. The cells were cultured until macroscopically visible colonies developed in the wells. Once colonies were observed, the cell culture was terminated, and each well was fixed using a 4% PFA solution, stained with 0.1% crystal violet, and counted using ImageJ software.
Wound healing assay
Cells were treated with a 0.25% trypsin solution (containing EDTA) and seeded in 6-well plates at a density of 5.0 × 105 cells/mL, with 1 mL per well, followed by gentle shaking to ensure even distribution. After 24 h, the control group was provided with serum-reduced medium, and the treatment groups were exposed to serum-reduced medium containing norcantharidin at concentrations of 15 μM, 30 μM, and 60 μM for 48 h. Photos were taken at 0, 12, 24, and 48 h intervals, while wound area calculations were conducted using ImageJ software.
Transwell migration and invasion assay
For Transwell migration assay, cells were treated with a 0.25% trypsin solution (containing EDTA) and seeded at a density of 3.0 × 105 cells/mL and 100 μL per well in the upper chamber of the Transwell. Serum-free medium (Control group), or serum-free medium containing norcantharidin at concentrations of 15 μM, 30 μM, and 60 μM were added to the upper chamber. Complete medium was added to the lower chamber, and the cells were cultured for 24 h. Finally, cells were fixed with 4% PFA, stained with 0.1% crystal violet staining solution, photographed, and counted using ImageJ software.
For Transwell invasion assay, 60 μL of Matrigel dilution (Matrigel:PBS = 1:8) was added to the upper chamber of the Transwell and placed in the incubator for gel formation. Then, 100 μL of serum-free medium was added to the upper chamber. Cells were digested with a 0.25% trypsin solution (containing EDTA) and seeded at a density of 3.0 × 105 cells/mL with 100 μL per well in the upper chamber. Subsequently, serum-free medium (Control group) or serum-free medium containing norcantharidin at concentrations of 15 μM, 30 μM, and 60 μM were added to the upper chamber. The lower chamber was loaded with complete medium, and cells were cultured for an additional 24 h. Lastly, cells were fixed using 4% PFA, stained with 0.1% crystal violet staining solution, photographed, and counted with ImageJ software.
Cell cycle detection
Cells were treated with a 0.25% trypsin solution (containing EDTA) and seeded at a density of 3.0 × 105 cells/mL, with 1 mL per well in 6-well plates, and cultured for 24 h. Following this, the control group received complete medium, while treatment groups were exposed to complete medium with norcantharidin at concentrations of 15 μM, 30 μM, and 60 μM for an additional 48 h. Upon completion, cells were digested with 0.25% trypsin solution (without EDTA), and cell suspensions were collected, centrifuged at 1500 rpm for 5 min, fixed with pre-chilled anhydrous ethanol, and incubated at − 20 °C for 24 h. Cells were resuspended in PBS and processed using the PI staining solution according to kit instructions. Red fluorescence was detected using flow cytometry at an excitation wavelength of 488 nm, and data were analyzed using FlowJo software.
Cell apoptosis detection
Cells were treated with a 0.25% trypsin solution (containing EDTA) and seeded at a density of 3.0 × 105 cells/mL, with 1 mL per well in 6-well plates and cultured for 24 h. the control group received complete medium, whereas treatment groups were cultured in complete medium containing norcantharidin at concentrations of 30 μM, 60 μM, and 90 μM for 48 h. After collecting the original culture medium and washing with PBS, cells were digested with 0.25% trypsin solution (without EDTA) and suspensions were collected. Following centrifugation at 600g for 5 min, the supernatant was discarded, and cells were processed according to the instructions provided in the Annexin V-FITC/PI Fluorescence Double Staining Cell Apoptosis Detection Kit. Immediate flow cytometry analysis was performed, and data were analyzed using FlowJo software.
Transcriptome sequencing and data quality control
PE libraries were prepared following the instructions provided in the mRNA-seq Lib Prep Kit. mRNA was isolated from total RNA using oligo(dT) magnetic beads and fragmented in Abclonal First Strand Synthesis Reaction Buffer. Subsequently, cDNA synthesis was performed using mRNA as a template, and the double-stranded cDNA fragments were ligated to adapter sequences. Library fragments were amplified using PCR, with library quality assessed via Agilent Bioanalyzer 4150. Alkaline denaturation transformed them into single-stranded libraries, followed by paired-end sequencing on the NovaSeq 6000 platform.
FastQC 0.11.9 software assessed raw data quality, with Trim_galore 0.6.6 software removing adapter sequences and filtering low-quality data (bases with quality scores ≤ 25 accounting for over 60% of reads) and reads with an N ratio > 5%. This procedure yielded Clean Reads for subsequent analysis. Clean Reads were aligned to reference genome data using HISAT2 software, generating Mapped Reads for further investigation. The FeatureCounts tool calculated the read counts for each gene, with FPKM values determined based on gene length.
DEGs expression analysis and enrichment analysis
Untreated HGC-27 cells served as the control group, while HGC-27 cells treated with norcantharidin constituted the treatment group. The DESeq2 package in R software was employed for inter-group DEGs expression analysis, with screening criteria set as |log2FoldChange|> 1 and p-value > 0.05 to identify DEGs before and after norcantharidin intervention on GC.
DEGs, downregulated DEGs, and upregulated DEGs were analyzed separately for biological processes (BP), cellular components (CC), molecular functions (MF), and KEGG pathways using the David database (https://david.ncifcrf.gov/), GO database (http://geneontology.org/), and KEGG database (http://www.kegg.jp). The hypergeometric distribution algorithm calculated the significance of DEGs within corresponding GO terms and KEGG pathways. Entries with p-value < 0.05 were selected for analysis of their biological significance and were used for subsequent key gene identification.
Key target acquisition
Jvenn (http://www.bioinformatics.com.cn/static/others/jvenn/) online plotting tool was utilized to obtain the intersection of norcantharidin upregulated, downregulated GC DEGs, and the DEGs in GC patients from the TCGA-STAD dataset [19]. These intersections were subsequently defined as key molecules in norcantharidin treatment of GC.
Gene set enrichment analysis was performed on the key molecules from norcantharidin intervention in GC using the Hallmark gene set from the MsigDB database (https://www.gsea-msigdb.org/gsea/msigdb) [20,21,22]. The hypergeometric distribution algorithm calculated the enrichment significance of each DEGs within the Hallmark gene set, with entries featuring a p-value < 0.05 used for selecting the core molecules.
Using the STRING database, the PPI network of key molecules involved in norcantharidin intervention in GC was constructed, setting the required minimum interaction score = 0.4. This network was imported into Cytoscape software for PPI network visualization, with the Cytohubba plugin revealing the key modules and core molecules within the network [23, 24]. Appropriate gene sets were subsequently selected based on enrichment analysis results; intersections were used to screen for key targets.









留言 (0)