
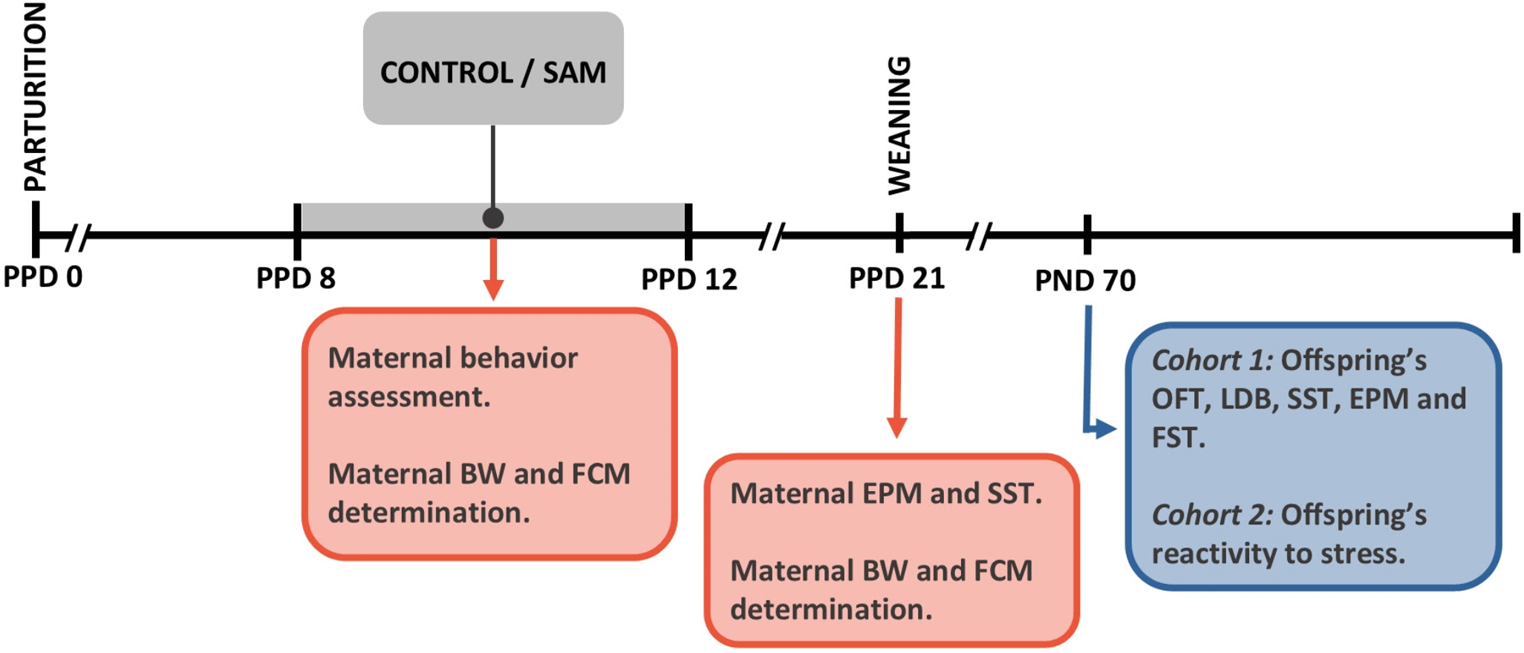
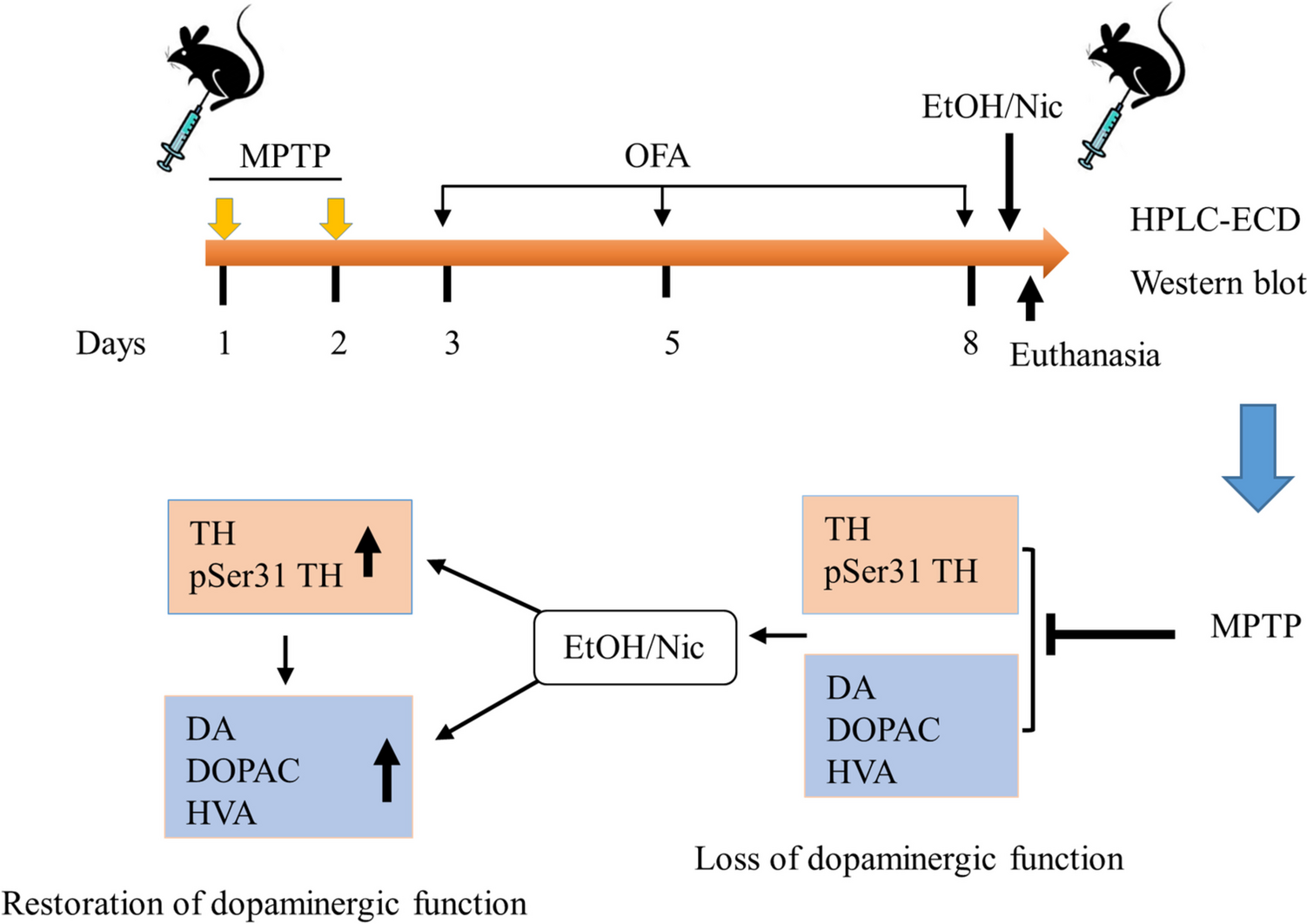
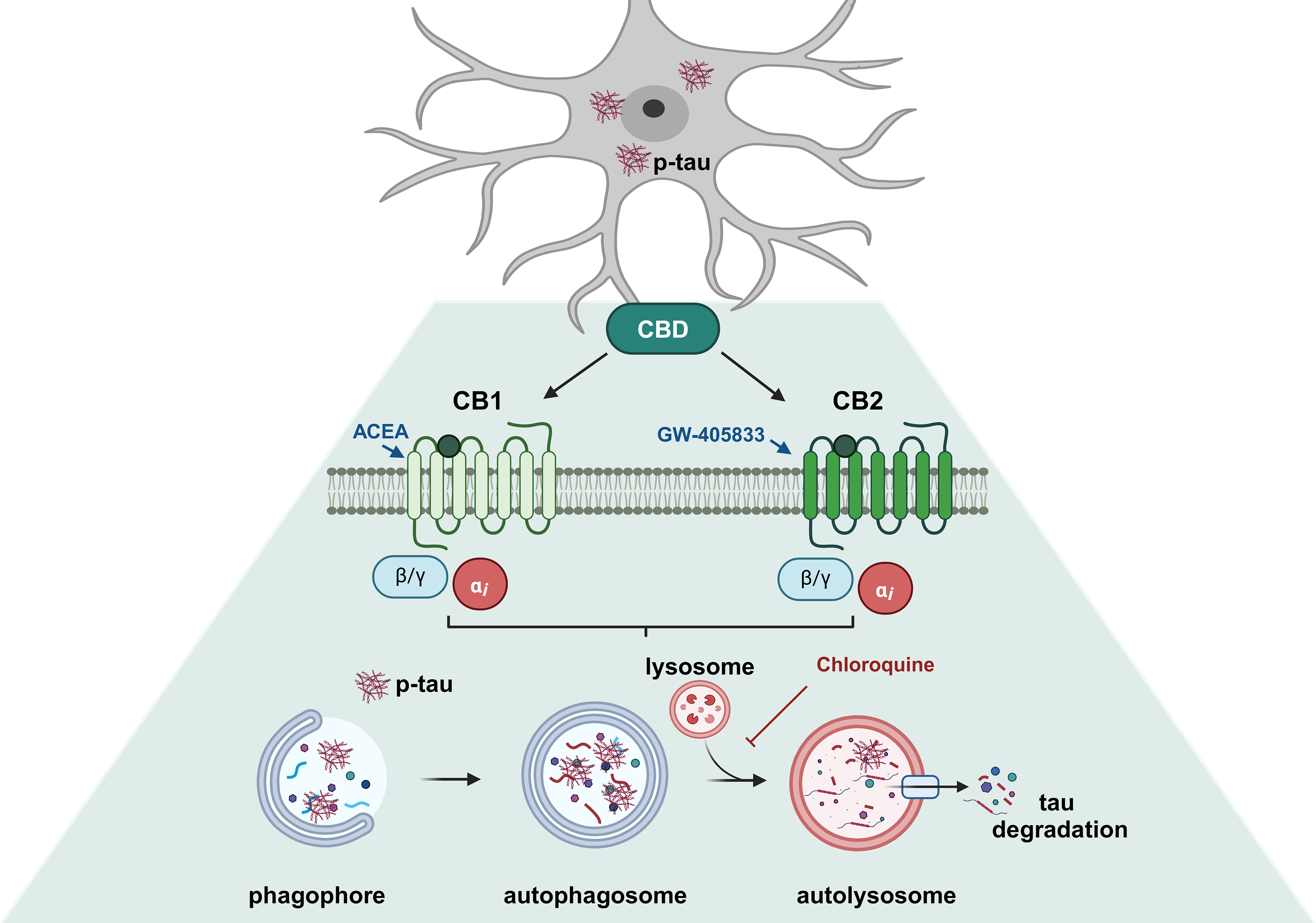
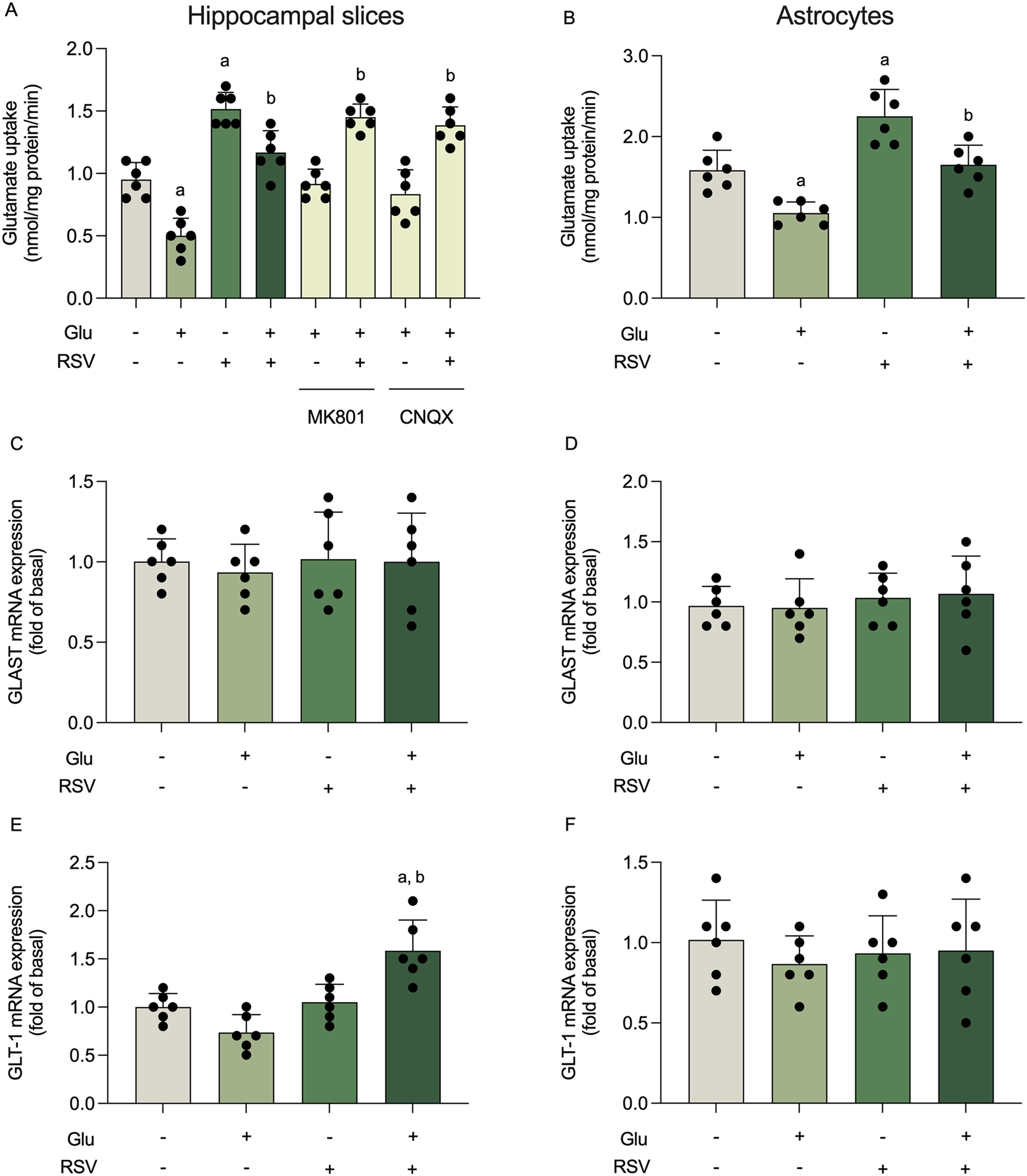
記住我



The goal of this study was to evaluate if A1AR stimulation is the mechanistic driver of the A/N effect of ENBA. Therefore, our focus was the pharmacologic actions of ENBA on prevention and termination of seizures, mitigation of brain pathology, and enhancement of survival.
Fig. 4
EEG gamma power and spike frequency analysis following soman (GD) exposure and treatments. Recording was analyzed using QuoStatus (Python-based software, University of Utah) to calculate gamma band power (4A) and spike frequency (4B). KIKO mice were pretreated with HI-6 (125 mg/kg, i,p,) 30 min prior to challenge with a dose of GD (33 µg/kg, s.c) and treated one min later with atropine methyl nitrate (4 mg/kg, i.p.). Animals were randomly assigned to one of the 4 treatment groups: AP (atropine sulfate + 2-PAM), MDZ (AP + midazolam), ENBA (AP + ENBA), or MDZ + ENBA (AP + midazolam + ENBA). Treatments were administrated i.p. at 15 min after GD-induced EEG seizure onset. EEG activity was analyzed for the first 5 h following GD exposure using REMLS/Mixed effect model and Dunn’s multiple comparisons. (*) indicates significantly different (p ≤ 0.05) from AP group. Significant reductions in both power (4A) and spike frequency (4B) were prominent within 15 min of treatment ([AP vs. ENBA: Power: p < 0.001 and Spike Frequency: p < 0.001] and [AP vs. ENBA + MDZ: Power: p = 0.016 and Spike Frequency: p < 0.001]). Robust control of EEG spiking in the gamma power band was maintained only by ENBA-included treatment groups throughout the rest of the recording, confirming no anti-seizure effects from the AP and AP + MDZ treatments
Table 2 Efficacy of medical treatments with and without ENBA following saline and soman (GD) exposureA typical example of EEG tracings of GD-induced SSE and ENBA or MDZ treatment in KIKO mice is shown in Fig. 3. Fifteen min after GD-induced SSE onset, animals were treated with either ENBA or MDZ (i.e., AP + ENBA or AP + MDZ treatment group, respectively). The top tracing is the baseline brain EEG activity; second tracing shows the onset of status epilepticus; third tracing displays the SSE; the 4th shows treatment with ENBA and termination of SSE with the tracing gradually turning into an isoelectric state; the 5th and 6th tracings display the continuing isoelectric activity at 5 h and 24 h, respectively, after ENBA treatment. The 7th and 8th tracings show EEG seizure activities were unabated at 5 h and 24 h, respectively, after MDZ (i.e., AP + MDZ) treatment, which are similar to previous observations with rats and guinea pigs; the seizure activities were still visible at 24–48 h after GD exposure in the survivors, even though the frequency and spike amplitude were much reduced (McDonough and Shih 1993, 1997).
Table 2 shows the effects of treatment groups on latency to isoelectric state, response fraction of non-isoelectric EEG at 24 h, time to seizure suppression, response fraction of seizure return, ENBA’s duration of action, and neuropathology scores in survivors at 14 days. In animals subjected to saline control exposure, groups of mice treated with ENBA and MDZ + ENBA produced a very rapid suppression of normal EEG activity to an isoelectric state within minutes with an average of 1.98 ± 0.40 and 1.60 ± 0.40 min, respectively. Furthermore, this effect lasted for 24 h after treatment; no single animal presented with normal baseline-like EEG activity within 24 h. The latency to isoelectric state and ENBA’s duration of action for all 16 saline-exposed ENBA-treated mice were within seconds of each other demonstrating ENBA at 10 mg/kg was having the same impact on EEG, with and without MDZ. On the other hand, for the groups treated with AP and AP + MDZ (= standard MCM), their EEG activity did not deviate much from baseline and were not sedated throughout the 24 h period after treatment. None of these 4 treatment groups following saline control exposure caused any visible neuropathology and all animals survived 14 days after saline exposure and treatments.
In mice exposed to GD, treatment with ENBA and MDZ + ENBA at 15 min after SSE onset induced a very rapid suppression of ongoing SSE activity (3.06 ± 1.11 and 2.00 ± 1.88 min, respectively; Table 2). Furthermore, this promoted an isoelectric state almost immediately after and lasted for the 24 h after treatment. With and without MDZ, all 20 mice presented without seizure reoccurrence at 24 h, demonstrating no difference on EEG between these two ENBA-treated groups in terms of time to seizure suppression and ENBA’s duration of action. Furthermore, neither of these two ENBA-included treatment groups displayed any visible neuropathology at 14 days after GD exposure and treatments, even though they endured at least 15 min of continuing SSE seizure activity before treatment was given. On the other hand, for the groups treated with AP and AP + MDZ, their ongoing SSE seizure activity continued for more than 24 h and subsequently exhibited severe neuropathology (~13 to 14 out of 24 maximum scores) in survivors at 14 days.
It is interesting to note that the effects of ENBA, in either ENBA alone or MDZ + ENBA treatment groups, on the latency to isoelectric state and seizure suppression under saline and GD exposure, respectively, and on the duration of EEG suppression under both saline and GD exposures are identical. This indicates that the action of ENBA is not influenced by the state of brain activities, whether it’s normal (saline exposure) or under severe ongoing SSE activity (GD exposure). Furthermore, under 15 min of severe and intense CNS seizure activity after GD intoxication, treatment with an MED of ENBA relieved neurons from pathological consequences observed at 14 days after such exposure.
Figure 4 displays the EEG gamma power band (Fig. 4A) and spike frequency (Fig. 4B) analysis during the first 5 h following GD exposure and treatments on the experimental day. The EEG reflects changes in seizure intensity after GD and treatment. Recording was analyzed using QuoStatus (Python-based software, University of Utah) to calculate gamma band power and spike frequency. All groups were not significantly different at baseline for power (range = 0.18 ± 0.30 to 0.41 ± 0.33 dB; AP vs. MDZ: DF = 17.7, P = 0.469; AP vs. ENBA: DF = 17, P = 0.329; AP vs. MDZ + ENBA: DF = 16.9, P = 0.455) and spike frequency measurements (range = 2.62 ± 5.35 to 0.25 ± 0.59 Hz; AP vs. MDZ: DF = 14.1, P = 0.977; AP vs. ENBA: DF = 8.15, P = > 0.999; AP vs. MDZ + ENBA: DF = 8.2, P = 0.451). Within the AP group, power ranged from 3.79 ± 0.52 to 1.57 ± 0.70 dB and spike frequency ranged from 25.87 ± 3.18 to 3.40 ± 1.91 Hz for the 5 h after GD exposure. Power was significantly above baseline for all 5 h (1 h: DF = 9, P = < 0.001; 5 h: DF = 9, P = 0.049). Spike frequency was also significantly above baseline for all 5 h (1 h: DF = 8, P = < 0.001; 5 h: DF = 8, P = 0.005) AP failed to reduce the SSE activity on EEG after GD. Within the MDZ group, power ranged from 3.40 ± 0.45 to 0.89 ± 0.45 dB and spike frequency ranged from 24.42 ± 5.74 to 1.18 ± 3.33 Hz throughout the 5 h after GD. MDZ power and spike frequency were significantly above baseline for 1.5 h after GD (power at 1.5 h: DF = 9, P = < 0.001; spike frequency at 1.5 h: DF = 9, P = 0.008) then began ramping down afterwards. Within the ENBA group, mean power reduced rapidly from 2.17 ± 0.51 at 15 min of treatment to -4.91 ± 0.28 dB within 45 min after treatment, while change in mean spike frequency dropped to -20.00 ± 5.05 Hz within 15 min of treatment. This overall reduction effect on EEG activity sustained throughout the 5 h after GD. Power was significantly reduced compared to baseline beginning at 0.5 h after GD (− 4.91 ± 0.28 dB; DF = 8, P = < 0.001). Spike frequency was significantly below baseline 0.5 h after GD (− 20.00 ± 5.05 Hz; DF = 8, P = < 0.001) and maintained below this throughout the rest of the initial experimental day, (5 h: − 26.65 ± 4.42 Hz). Within the MDZ + ENBA group, the impact on EEG was similar in trend to ENBA alone group. The mean power reduced from 1.62 ± 1.78 at 15 min of treatment to -5.71 ± 0.28 dB within 45 min after treatment which was significantly reduced compared to baseline (or 1 h after GD: − 5.71 ± 0.41 dB, DF = 8, P = < 0.001). The mean spike frequency was significantly below baseline at 15 min after treatment (or 0.5 h after GD: − 20.75 ± 9.15 Hz, DF = 8, P = < 0.001) and reduced further to -26.70 ± 5.85 Hz from 1 h and sustained throughout the 5 h after GD. Within 15 min of treatment, significant differences in change for ENBA and MDZ + ENBA against the control (AP group) were evident for both power (AP vs. ENBA: DF = 16.9, P = < 0.001; AP vs. MDZ + ENBA: DF = 9.25, P = 0.016; Fig. 4A) and spike frequency (AP vs. ENBA: DF = 13.2, P = < 0.001; AP vs. MDZ + ENBA: DF = 9.74, P = < 0.001; Fig. 4B).
Overall, the MDZ treatment intermittently reduced power significantly compared to the control group (AP vs. MDZ at 1 h: DF = 13.4, P = 0.037; 2 h: DF = 17.9, P = 0.027, and 4.5 h: DF = 17.5, P = 0.013). The effect on spike frequency with MDZ was similar with a short reduction lasting approximately 1 h when compared to the AP control group (AP vs. MDZ at 1.5 h: DF = 16, P = 0.003; and 2.5 h: DF = 12.5, P = 0.022). However, only the ENBA and MDZ + ENBA treatments resulted in a lasting reduction in mean power throughout the 5 h (AP vs. ENBA at 5 h: DF = 11.9, P = < 0.001; AP vs. MDZ + ENBA at 5 h: DF = 16, P = < 0.001). Changes in the mean spike frequency were also lasting throughout the 5 h (AP vs. ENBA at 5 h: DF = 10.7, P = < 0.001; AP vs. MDZ + ENBA at 5 h: DF = 9.54, P = < 0.001). The ENBA alone or in combination to MDZ, effectively controlled the SSE-dependent rise in power and spike frequency compared to AP.
Neuropathological damage was assessed by a trained veterinary pathologist who was unaware of treatment paradigm and scores determined for each of the six brain regions (amygdala, cerebral cortex, piriform cortex, thalamus, dorsal hippocampus, and ventral hippocampus) by the percentage of degeneration and necrosis of neurons in each region with a maximum total severity score of 24 (McDonough et al. 1995; Loughery et al. 2021; Shih 2023). Other neuropathologic changes such as neuropil vacuolation and gliosis were noted in the GD-exposed mice but not used to determine the damage severity. Generally, animals with neuropathological damage in one region had damage in all regions, though the hippocampus was spared in some instances. The degree of damage across regions varied. For all of the survivors at 24 h, no brain pathology occurred when their seizure activity was terminated by the treatment of ENBA (Table 2). For example, ENBA treatment at 15 min after SSE onset showed a total protection in neuropathology score (0.0 ± 0.0) compared to AP- and AP + MDZ-treated groups (13.71 ± 5.65 [7] and 14.20 ± 2.68 [5], respectively). In the latter two treatment groups, brain sections have multifocally extensive neuronal necrosis and degeneration with neuropil vacuolation, while in the two ENBA-included groups brain sections have undamaged neurons with multifocal areas of ‘dark neurons’ (a handling artifact). The detailed examples of H&E-stained histology images in 6 brain regions of KIKO mouse induced by GD exposure and protected by treatment with ENBA alone have been published elsewhere (see Shih 2023 for details).
GD Toxicity Effects, Lethality and Quality-of-Life. (Tables 2, 3, 4, and 5, Supplemental Table 20)For saline-exposed groups, there was no mortality at 24 h following any of the 4 treatments; thus, the KIKO mouse is able to tolerate doses of ENBA and MDZ + ENBA, even though they produced a very rapid suppression of normal EEG activity to an isoelectric state within min and the effect lasted for 24 h. We had observed earlier, in our MED dose determination study, that ENBA up to 45 mg/kg in KIKO mouse did not cause mortality in 24-h (Shih 2023).
Table 3 Quality of health after soman (GD) exposure and treatmentTable 4 Toxicity index after soman (GD) exposureTable 5 Quality of life at 14 days after soman exposure and treatmentFor GD exposed groups, after AP, AP + MDZ, AP + ENBA and AP + MDZ + ENBA treatments (10 mice per group), the lethality at 24 h was 3, 2, 1, and 1, respectively, and at 14 days progressed to a final total of 3, 5, 2, and 1, respectively (Table 2). A probability assessment on time and survival determined no difference in mean time of survival between the treatment curves. Death events were clustered around 24 h. Additionally, animals who were dependent on supportive post-exposure care due to a plateau in recovery were observed and further noted as survival with abnormality. To assess the interaction of survival event with abnormalities (e.g., poor health), a measure of effects associated with GD toxicity was done. Specifically, a plot of daily assessments (temperature and body weight) was used to calculate the variability in severity across time and to capture the index of toxic effect across all treatments with higher confidence, when compared to a probability assessment of binomial event outcomes.
The quality of health was summarized for temperature and body weight by counting total observations and identifying the sum of poor observations (indicative of poor health) and expressed as ratios for each treatment group (Table 3). A temperature at or below 35 °C and -7% weight loss from baseline or greater were thresholds indicative of poor health. For the AP (control) group, 8% of the temperature and 31% of the weight observations were below threshold, of those observations the scatter was throughout the 14 days after treatment. This reflects variable recovery after GD. The MDZ group had high percentages of poor health observations in both measures (temperature = 57% and weight = 94%) dispersed throughout the 14 days. On the other hand, ENBA and MDZ + ENBA had minimal poor observations in both measures (temperature = 11–12% and weight = 11%). The observations were clustered between day 1–2, which aligns to the recovery period of A1AR agonist-induced sedation and hypothermia.
The level of toxicity after GD exposure was indexed across all 40 GD-exposed animals (Table 4). This quantifies the toxic effect and orders the level of toxicity as such; absence of toxic effect = 0%, negligeable toxic effect = 25%, median toxic effect = 50%, high impact toxic effect = 75%, gross toxic effect = 100%. The distance between average rank per group measures the toxicity variability for each treatment group. Consequently, as toxic effect increased over time (remained unmitigated), the risk of lethality over time also increased. Across all GD-exposed animals, 12 of the 40 had an absence of toxicity after treatment (0%), 6 of the 40 had inconsequential effects (25%), 6 had median to high impact toxic effects (50–75%), and 16 of the 40 were grossly impacted (100%). Next, we reviewed the distribution within the levels of toxicity by treatment group membership. The AP group had mice distributed between the upper 4 levels of toxicity (25–100%). Of the 10 mice belonging to the AP group, 5 were at the 50–75% levels and 4 were at the 100% level. Even more severe, the MDZ group had mice distributed between the upper 2 levels of toxicity (75–100%). Of the 10 MDZ-treated mice, 1 was at 75% while 9 were at the 100% level. In contrast, the ENBA and MDZ + ENBA groups had mice distributed primarily between the lower 2 levels of toxicity (0–25%). The ENBA group had 8 mice at 0–25%, and 2 at 100% who died around 24 h. The MDZ + ENBA group was similar, with 9 mice at 0–25% and 1 at 100% who died early on.
At 14-days, the quality-of-life (QoL) measures indicated that the control group treated with AP had 2 out of 7 animals present with high levels of toxicity, and MDZ had 4 out of 5 animals display high levels of toxicity (Table 5). While none of the 8 or 9 survivors in either ENBA or MDZ + ENBA groups had animals in such a weak condition. For the latter two ENBA-treated groups, the surviving animals were all self-sustainable and did not require extra post-exposure food, heat, and fluid support at the end point of the study.
Effects on Body Temperatures (Fig. 5; Supplemental Fig. 1; Supplemental Tables 9–11)Fig. 5
Fourteen-day body temperature recordings following saline (sham) or soman (GD) exposure and treatments. KIKO mice were treated with HI-6 (125 mg/kg, i,p,) 30 min prior to challenge with a dose of saline (5A) or GD (33 µg/kg, s.c.; 5B) and treated one min later with atropine methyl nitrate (2 mg/kg for saline-exposed, 4 mg/kg for GD-exposed, i.p.). Animals were randomly assigned to one of the 4 treatment groups: AP (atropine sulfate + 2-PAM), MDZ (AP + midazolam), ENBA (AP + ENBA), or MDZ + ENBA (AP + midazolam + ENBA). Treatments were administrated i.p. at 15 min after GD-induced EEG seizure onset or relevant time after saline exposure groups. Body temperature was recorded and statistically compared for the 5 h during experimental day, at 24 h, and on day 7 and 14 following exposure. Temperature was analyzed using REMLS/Mixed effect model and Dunn’s multiple comparisons for within and between group comparisons within each exposure. (*) indicates significantly different (p ≤ 0.05) from respective AP treatment group. ENBA treatments affect temperature profoundly within 4 min of treatment in saline-exposed mice (5A) compared to AP-treated mice ([AP vs. ENBA: p = 0.016] and [AP vs. ENBA + MDZ: p = 0.005]) up to 24 h after administration (AP vs. ENBA and ENBA + MDZ: p < 0.001); however, this was reversible with the addition of short-term thermal support at the 24-h mark. Similarly, ENBA profoundly impacted temperature in GD-exposed mice compared to AP-treated mice (5B) shortly after treatment ([at 4 min, AP vs. ENBA, p = 0.029] and [at 8 min, AP vs. MDZ + ENBA, p = 0.062] for up to 24 h after administration (AP vs. ENBA and ENBA + MDZ: p < 0.001)
Since A1AR agonists induce pharmacologic effects on various bodily physiological functions (see review by Borea et al. 2018; Effendi et al. 2020), the following are a collection of the actions of ENBA we have observed in association with our main objective of investigating ENBA’s neuroprotective effects following GD exposure.
Beyond suppression of neuronal activity in the CNS, A1AR agonists had significant peripheral effects, such as hypothermia (Carlin et al. 2017). We defined the hypothermic body temperature range at 32.0 ± 2.0 °C in our studies. All ENBA-included treatments with and without GD exposure induced some degree of hypothermia, bradycardia (i.e., reduction of heart rate), and sedation in rats (Thomas et al. 2019; Loughery et al. 2021). Similar effects are observed in the KIKO mice following ENBA treatment. In saline exposure (Fig. 5A), mean baseline body temperatures ranged between 36.9 ± 0.7 °C to 37.4 ± 0.4 °C across all treatment groups. After AP (control) treatment, there was no appreciable change in body temperature from baseline throughout the study (mean range was 36.8 ± 0.7 °C to 36.0 ± 0.6 °C). The MDZ group’s mean body temperatures ranged from 35.0 ± 1.5 °C to 36.3 ± 0.6 °C after treatment. Within the MDZ group, treatment induced a significant reduced body temperature from baseline beginning at 1 h (35.1 ± 1.2 °C; DF = 7, P = 0.034), through 4 h after treatment (35.8 ± 0.6 °C; DF = 7, P = 0.004). When compared directly against the control (AP group), MDZ had significantly lower body temperatures intermittently after treatment (Saline/AP vs. MDZ: 15 min (DF = 11.1, P = 0.031); at 30 min (DF = 10.9, P = 0.035); and at 3 h (DF = 13.5, P = 0.027). Mean body temperatures for MDZ corrected by 4 h after treatment (35.8 ± 0.6 °C), and were comparable to AP. On the other hand, the reduction of temperature by ENBA (10 mg/kg, i.p.) was large and prevalent for 24 h. The rate of change for body temperature within the first h after ENBA was -0.13 °C per min and for MDZ + ENBA was -0.15 °C per min. After only 4 min, ENBA administration reduced body temperature significantly compared to baseline (ENBA: 35.7 ± 0.7 °C (DF = 7, P = 0.018); MDZ + ENBA: 35.5 ± 0.7 °C (DF = 7, P = 0.042)). ENBA and MDZ + ENBA groups sustained the hypothermic effect throughout the 5 h after treatment. They presented with a baseline of 37.4 ± 0.4 °C (8) for ENBA and 36.9 ± 0.7 °C (8) for MDZ + ENBA then reducing to 21.5 ± 0.6 °C and 21.5 ± 0.9 °C, respectively, at 5 h (300 min) after treatment, or a reduction of 42% from baseline. These groups continued to experience a decrease in body temperature overnight; at 24 h after treatment, temperatures remained severely hypothermic compared to baseline (ENBA: 20.5 ± 0.7 °C (DF = 7, P = < 0.001); MDZ + ENBA: 21.1 ± 1.0 °C (DF = 7, P = < 0.001). However, by 48 h, the temperatures all returned to a normal range and maintained as such to the end of 14 days. There was no apparent difference between the rate of change in these two ENBA-treated groups overall, showing that MDZ, when part of the treatment regimen, had no added effect. Thus, the body temperature records showed that ENBA (10 mg/kg) induced significant hypothermia that lasted for about a day under saline control exposure condition.
For mice exposed to GD (Fig. 5B), mean baseline body temperatures were similar for all groups. Specifically, 37.3 ± 0.6 °C (10) for AP, 37.4 ± 0.4 °C (10) for MDZ, 37.7 ± 0.4 °C (10) for ENBA, and 37.6 ± 0.4 °C (10) for MDZ + ENBA. After exposure, temperatures ranged between 34.7 ± 0.5 °C to 35.2 ± 0.5 °C at 15 min after SSE onset. All groups were significantly below baseline before treatment (vs. 15 min PE: AP (DF = 9, P = < 0.001); MDZ (DF = 10, P = < 0.001); ENBA (DF = 9, P = < 0.001); and MDZ + ENBA (DF = 9, P = < 0.001)). In addition, all groups were not significantly different before treatment administration, when compared to the control AP designated mice.
Within 4 min of treatment, an equivalent and significant reduction on body temperature was observed within the AP (control) and MDZ treatment groups (AP: 33.0 ± 1.6 °C (DF = 9, P = < 0.001); MDZ: 32.8 ± 2.0 °C (DF = 9, P = < 0.001)). They were not significantly different from each other throughout the 5 h while presenting with a reduction of ~ 12% from baseline body temperature, which then could be a factor attributed to the GD exposure. A Mann–Whitney U test was performed to compare average body temperature between the saline exposed AP-treated group to the GD exposed AP-treated group. The temperature was significantly different between the two after exposure, (8 min PE: Mann–Whitney U = 6.5, q-value = 0.0015). The same test was performed between the saline exposed MDZ-treated group to the GD exposed MDZ-treated group and had similar results after exposure, (8 min PE: Mann–Whitney U = 2, q-value = 0.00016). Across exposures for both treatment groups, similar trends were observed regardless of AP or MDZ treatment (Supplemental Table 11), showing that GD has a physiologically relevant impact on body temperature overall. By the 24 h timepoint, AP had mean body temperatures comparable to baseline (35.9 ± 2.3 °C), but the MDZ group, compared to baseline, was hypothermic (32.1 ± 3.9 °C; DF = 8, P = < 0.001). After 24 h observations, animals (regardless of treatment) who were at or below 35 °C were placed on the heat pad in the animal holding room and monitored daily. Animals were taken off heat support when temperatures were above 35.1 °C and had regained mobility. The body temperature of the AP group returned to 37.0 ± 1.3 °C on the second day on average. However, the AP + MDZ group remained between the range of 32.1 ± 3.9 °C to 32.3 ± 0.0 °C for the next 3 days before returning to 36.8 ± 0.5 °C on the 4th day after GD exposure. Thus, GD exposure might have extended the duration of mild hypothermia by MDZ when compared to the saline exposure groups.
On the other hand, the administration of ENBA (15 mg/kg, i.p.) and MDZ + ENBA treatment caused body temperature to reduce by over 10 °C from normal temperature ranges over 2 h. The rate of change for body temperature within the first h after ENBA was -0.13 °C per min and for MDZ + ENBA was -0.13 °C per min. At 15 min after GD and before treatment, body temperature was already reduced significantly to 34.7 ± 0.5 °C for ENBA designated mice (vs. baseline: DF = 9, P = < 0.001) and 35.2 ± 0.5 °C for MDZ + ENBA designated mice (vs. baseline: DF = 9, P = < 0.001). After only 4 min, body temperature dropped further compared to group baseline (ENBA: 33.5 ± 0.6 °C (DF = 9, P = < 0.001); MDZ + ENBA: 34.0 ± 0.7 °C (DF = 9, P = < 0.001)).
When compared changes in body temperature against the control AP group, the ENBA group presented with a significant difference at 4 min after treatment (ENBA: 33.5 ± 0.6 °C (DF = 16.1, P = 0.029)). After 15 min from treatment, both ENBA-treated groups were significantly lower compared to AP (ENBA: 31.0 ± 0.7 °C (DF = 13.3, P = < 0.001); MDZ + ENBA: 31.8 ± 0.5 °C (DF = 11.2, P = < 0.001).
At 5 h after treatment, both ENBA-treated groups had a severe reduction of ~ 42% from baseline in body temperatures with ENBA at 22.4 ± 3.7 °C (DF = 8, P = < 0.001) and MDZ + ENBA at 21.2 ± 0.9 °C (DF = 9, P = < 0.001). These two groups sustained the hypothermic body temperature overnight; the 24-h average temperatures were 23.6 ± 3.6 °C and 22.3 ± 3.4 °C, respectively. By 48 h, temperatures returned to a normal range of 38.4 ± 0.8 °C and 37.7 ± 0.8 °C, respectively, and were maintained as such to the end of 14 days. There was no difference between the rate of change for ENBA and MDZ + ENBA treatments, indicating that ENBA was the main factor in the cause of hypothermia. The data showed that ENBA (15 mg/kg) induced a marked reduction of body temperature that lasted for 24 h following GD exposure and 15 min of ongoing SSE.
When comparing the average body temperature between the saline exposed ENBA-treated group to the GD exposed ENBA-treated group, temperature was significantly different between the two after exposure, (4 min PE: Mann–Whitney U = 0, q-value = 0.00015) until 1 h due to the effect of GD on temperature significantly reducing temperature before treatment administration. However, they were no longer different by 2 h after treatment. This difference in temperature effect from agent was shorter between the saline exposed MDZ + ENBA group to the GD exposed MDZ + ENBA group, where by 8 min after treatment temperature was no longer significantly different between the two.
Effects on Heart Rates (Fig. 6; Supplemental Fig. 2; Supplemental Table 12–15)Fig. 6
Fourteen-day percent change in heart rate (HR) measurements following saline (sham) or soman (GD) exposure and treatments. KIKO mice were treated with HI-6 (125 mg/kg, i,p,) 30 min prior to challenge with a dose of saline (6A) or GD (33 µg/kg, s.c.; 6B) and treated one min later with atropine methyl nitrate (2 mg/kg for saline-exposed, 4 mg/kg for GD-exposed, i.p.). Animals were randomly assigned to one of the 4 treatment groups: AP (atropine sulfate + 2-PAM), MDZ (AP + midazolam), ENBA (AP + ENBA), or MDZ + ENBA (AP + midazolam + ENBA). Treatments were administrated i.p. at 15 min after GD-induced EEG seizure onset or relevant time after saline exposure groups. Heart rate (HR) was recorded hourly on experimental day for 5 h, at 24 h, and on day 7 and 14 following exposure. The percent change from baseline was calculated as: (100 x (24 h HR – baseline HR))/baseline HR for each animal before averaging. HR was analyzed using REMLS/Mixed effect model and Dunn’s multiple comparisons for within and between group comparisons within each exposure. (*) indicates significantly different (p ≤ 0.05) from respective AP treatment group. ENBA and MDZ + ENBA negatively impacted HR within 1 h of treatment compared to AP treatment alone for saline (6A) and GD (6B) exposures (saline exposure: [AP vs. ENBA and MDZ + ENBA: p < 0.001]; GD exposure: [AP vs. ENBA: p = 0.003; AP vs. MDZ + ENBA: p = 0.002]). This effect was observed for 24 h after administration (saline exposure: [AP vs. ENBA and MDZ + ENBA: p < 0.001]; GD exposure: [AP vs. ENBA and MDZ + ENBA: p < 0.001]). The suppression of HR was readily reversible with gentle warming to overcome sedation after ENBA and HR readings were comparable to their respective AP treatment group by day 7
In addition to lowering body temperature, A1AR agonists rapidly reduced heart rate (HR). It is well known that adenosine reduces HR and impulse generation in heart tissues (Drury and Szent-Györgyi 1929; Headrick et al. 2011). At the start of the study, all KIKO mice HR averaged from 729 ± 41 to 770 ± 34 beats per min (BPM) at baseline. Within the AP group, there was no significant difference in percent change of HR from baseline 1 h after treatment. Within the MDZ group, treatment caused a significant reduction in HR from baseline from 1 h throughout the 5 h after treatment (vs. baseline at 1 h: DF = 7, P = 0.015; at 5 h: DF = 7, P = 0.003). Although throughout the saline exposure (Fig. 6A), the MDZ treatment group was not significantly different from the AP (control) group with mean HR change from baseline ranging from -11.4 ± 18.1% and -13.0 ± 8.2% (at h 1), to -8.8 ± 8.5% and -6.4 ± 3.0% (at h 5), for AP and MDZ respectively. Within the ENBA group, treatment induced a -78.5 ± 11.1% change from baseline by h 1 (DF = 7, P = < 0.001), while MDZ + ENBA treatment induced an even lower change of -86.8 ± 0.7% from baseline at h 1 (DF = 6, P = < 0.001). This induction of bradycardia was reliably observed within min of ENBA treatment, quickly accompanied by lethargy and pallor (Loughery et al. 2021). The depression of HR relative to baseline persisted to the 24-h endpoint for ENBA treatment alone or in combination with MDZ, reaching -82.5 ± 2.3% and -82.8 ± 4.6% of baseline HR (i.e., 132 ± 17 and 126 ± 33 BPM), respectively, after saline exposure with significantly lower rates compared to the control treatment (AP vs. ENBA at 24 h: DF = 11.9, P = < 0.001; AP vs. MDZ + ENBA at 24 h: DF = 13.1, P = < 0.001). The bradycardia was no longer detected at the 7th day measurement and beyond, both ENBA-treated groups were at or above average baseline HR. However, in the AP and AP + MDZ groups the HRs were relatively unchanged from baseline values.
For GD-exposed KIKO mice (Fig. 6B), the AP and MDZ treatment groups had HR equally impacted for 24 h after exposure, when comparing with saline-exposed groups. To compare across exposures, a Mann–Whitney U test was performed to compare the percent change in HR from baseline between the saline exposed AP-treated group to the GD exposed AP-treated group. The change was not significantly different between the two exposures after 1 h and remained comparable at 5 h. The same test was performed between the saline exposed MDZ-treated group to the GD exposed MDZ-treated group and there was no significant difference 1 h after treatment. A significant reduction began at 3 h after treatment and did not resolve by 5 h (3 h PT: Mann–Whitney U = 4, q-value = 0.0023; Supplemental Table 15). Within the AP group, a significant reduction in HR from baseline began at 1 h and persisted until 5 h after treatment, (-33.2 ± 30.0% Δ from baseline at 1 h: DF = 9, P = 0.036; to -18.5 ± 16.4% Δ from baseline at 5 h: DF = 9, P = < 0.001). Within the MDZ group, a similar reduction in HR from baseline began at 1 h and did not resolve 24 h after treatment, (-22.0 ± 17.3% Δ from baseline at 1 h: DF = 9, P = 0.016; -24.3 ± 17.3% Δ from baseline at 24 h: DF = 7, P = 0.022). Although, the change in HR of the MDZ treated group was not significantly different from the AP control group throughout the 24 h after exposure.
Similar to the effect observed within the saline exposure, ENBA and MDZ + ENBA treatment caused large reductions in HR from baseline. Presenting at 160 ± 72 BPM for ENBA and 148 ± 134 BPM for MDZ + ENBA at 1 h after treatment. This is a severe reduction from baseline for both groups, (-78.1 ± 9.4% Δ from baseline for ENBA at 1 h: DF = 7, P = < 0.001; − 80.7 ± 17.4% Δ from baseline for MDZ + ENBA at 1 h: DF = 7, P = < 0.001). HR maintained at this level for both groups throughout the 24 h period. When compared between exposures, treatment with ENBA or MDZ + ENBA did not present with a significant difference at any point (Supplemental Table 15). HR was measured again on the 7th day after exposure, where both ENBA-treated groups presented back to baseline range (Supplemental Table 14a).
Effects on Motor Activity and General State of Movement (Fig. 7, Table 6)Fig. 7
Fourteen-day motor activity (convulsions) recordings following saline (sham) or soman (GD) exposure and treatments. KIKO mice were pretreated with HI-6 (125 mg/kg, i,p,) 30 min prior to challenge with a dose of saline (
 ) or GD (33 µg/kg, s.c.;
) or GD (33 µg/kg, s.c.;
 ) and treated one min later with atropine methyl nitrate (2 mg/kg for saline-exposed, 4 mg/kg for GD-exposed, i.p.). Animals were randomly assigned to one of the 4 treatment groups: AP (atropine sulfate + 2-PAM; 7A), MDZ (AP + midazolam; 7B), ENBA (AP + ENBA; 7C), and MDZ + ENBA (AP + midazolam + ENBA; 7D). Treatments were administrated i.p. at 15 min after GD-induced EEG seizure onset or relevant time after saline exposure groups. Toxic motor activity was recorded and statistically compared for 5 h during experimental day, at 24 h, and on day 7 and 14 following exposure. Toxic motor score assessed physical seizure activity for severity (0 = none, 1 = fasciculations, 2 = localized tremors, 3 = full body convulsions). Treatment with ENBA and MDZ + ENBA equally reduced motor convulsions from a median score of 3 to a median score of 0 within 5 min after administration
) and treated one min later with atropine methyl nitrate (2 mg/kg for saline-exposed, 4 mg/kg for GD-exposed, i.p.). Animals were randomly assigned to one of the 4 treatment groups: AP (atropine sulfate + 2-PAM; 7A), MDZ (AP + midazolam; 7B), ENBA (AP + ENBA; 7C), and MDZ + ENBA (AP + midazolam + ENBA; 7D). Treatments were administrated i.p. at 15 min after GD-induced EEG seizure onset or relevant time after saline exposure groups. Toxic motor activity was recorded and statistically compared for 5 h during experimental day, at 24 h, and on day 7 and 14 following exposure. Toxic motor score assessed physical seizure activity for severity (0 = none, 1 = fasciculations, 2 = localized tremors, 3 = full body convulsions). Treatment with ENBA and MDZ + ENBA equally reduced motor convulsions from a median score of 3 to a median score of 0 within 5 min after administration
Treatments including ENBA produced rapid decrements of motor activity and subsequent sedation regardless of saline or GD exposure (Table 6 and Fig. 7). A functional observational battery (FOB), including assessments for startle reflex, righting reflex, motor activity, gait, and arousal was performed on each animal at regular intervals after saline and GD exposure and treatments (Tables 1 and 6). Of the various functional assessments, ambulation (e.g., general state of movement) was the most representative measure of function or incapacitation for these mice. That is, the ability to ambulate even slowly indicated the return of a righting reflex and at least a moderate degree of arousal or cognitive function. ENBA at 10 mg/kg in saline-exposed and 15 mg/kg in GD-exposed mice experienced severe immobility and depth of sedation; they became unconscious, which corresponded with the suppression of EEG activity. The suppression of EEG activity progressed rapidly within min after ENBA treatment (Table 2, Fig. 3). Subjects became unresponsive, displayed complete loss of muscle tension, and lacked a righting reflex. These effects lasted for more than 24 h and returned to baseline by the 7th day of observation time point. In saline-exposed mice (Fig. 7; green box), treatment with either AP (Fig. 7A) or AP + MDZ (Fig. 7B) showed no deficit in motor functions (score = 0, Table 6). However, in GD-exposed animals (Fig. 7, red box), these two treatment groups induced a higher degree of impaired movement and loss of muscle tension (scores ranges 1.2 – 3.0, Table 6). AP (Fig. 7A) and MDZ (Fig. 7B) treatments were not able to prevent or stop GD induced toxicity on muscular activity in these mice. They were prostrated and their muscular convulsions unabated. The most toxic effect of GD was the muscular convulsive activity, which was attenuated quickly and completely by the two ENBA treatment groups (Fig. 7C and D), clearly displaying the anticonvulsive efficacy of ENBA against GD-induced chronic and tonic muscular toxicity.
Table 6 Summary of Functional Observational Battey (FOB) scores for toxic signs after saline and soman (GD) exposures and treatmentsEffects on Body Weights (Table 7, Supplemental Table 16–20)Weight loss can indicate when an animal is under stress or suffering from poor health. At the start of the study, all KIKO mice body weights averaged from 26.3 ± 1.2 to 29.7 ± 1.4 g at baseline. Following saline exposure (Table 7) the AP (control) and MDZ treatment groups presented above baseline from day 2 throughout day 14. On day 7, the AP and MDZ groups had recovered weight above their day 1 average (+ 2.1 ± 2.2% Δ from baseline for AP: (DF = 56, P = < 0.001); + 0.2 ± 3.4% Δ from baseline for MDZ: (DF = 56, P = 0.001)). However, at day 2, the ENBA and MDZ + ENBA treatment groups presented -7.6 ± 3.0% and -7.7 ± 3.0% below baseline. This timepoint aligns to the recovery period from ENBA-dependent sedation and hypothermia. Therefore, these effects had an intermittent effect on normal behavior and weight. Weight recovered within 5% of baseline by day 4 for the ENBA group and continued to improve on day 7, presenting significantly improved compared to day 1 after exposure (-1.1 ± 2.1% Δ from baseline for ENBA: DF = 56, P = 0.009). The MDZ + ENBA group presented with the smallest change in weight after 24 h among the saline exposed groups. Their weight change was -0.975 ± 1.8% Δ from baseline on day 1 and was significantly abov
留言 (0)