
記住我
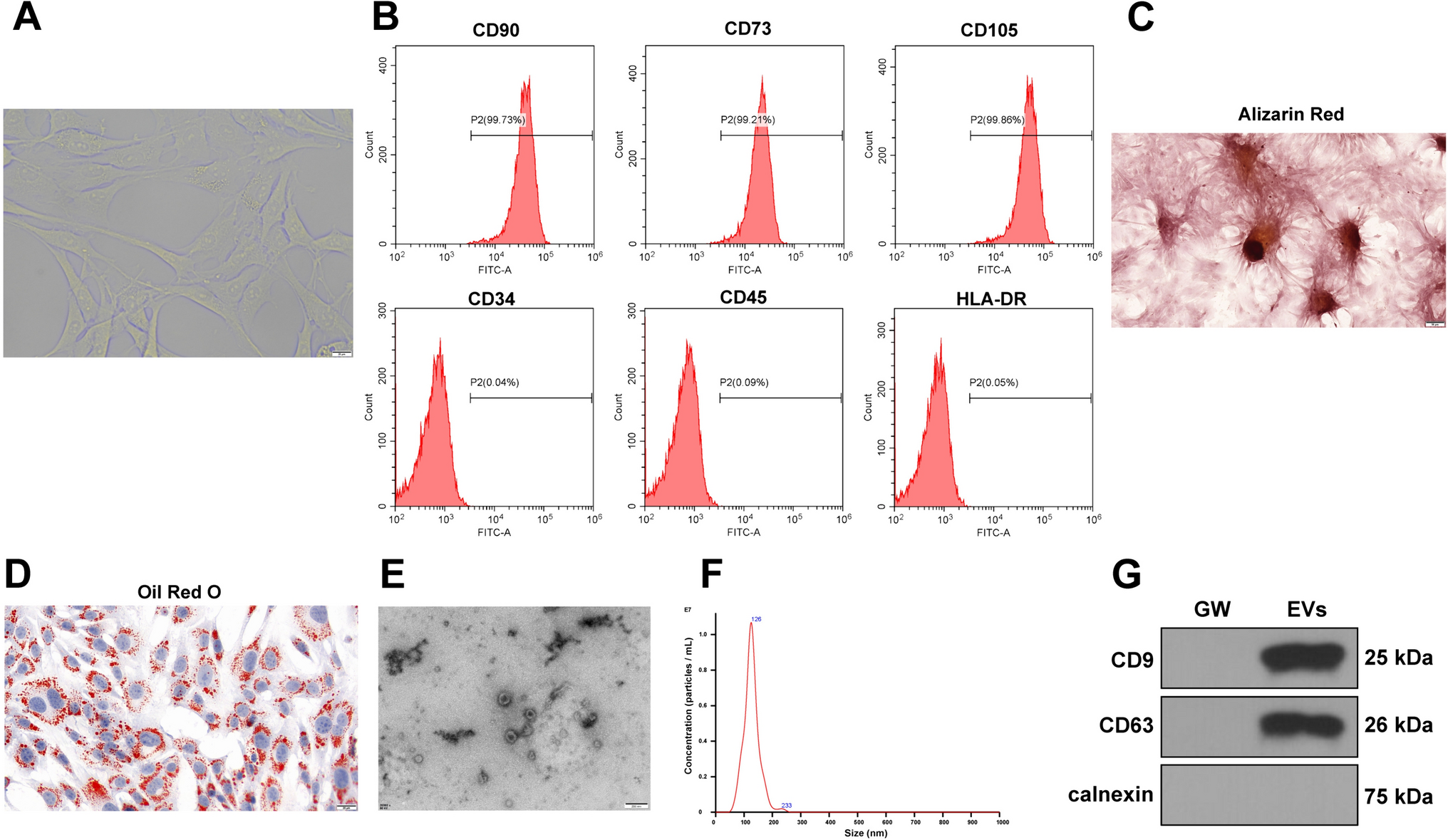





Human mesenchymal stem cells were isolated by plastic adherence from a whole bone marrow aspirate (Lonza, Gaithersburg, MD, USA) and cultured to passage 3 in basal DMEM containing 10% foetal calf serum and 1% penicillin–streptomycin (all Gibco, Waltham, MA, USA). MLO-A5 cells were kindly donated by Professor Linda Bonewald of the University of Missouri, Kansas. Cells were trypsinised and incorporated into a collagen hydrogel made from a stock solution of 9.81 mg/ml Type-I rat tail collagen (BD Biosciences, Berkshire, UK) and culture media in 24-well transwell culture inserts (Corning, Deeside, UK). The seeding conditions were 3 × 105 cells per hydrogel, 2.5 mg/ml final collagen concentration with a 300 µl initial volume. After 24 h, the cell-seeded hydrogels were transferred into 24-well plates containing 2 ml fresh osteogenic media (DMEM as above, plus ascorbic acid, 10−8 M dexamethasone and 2 mM sodium β-glycerophosphate, all Sigma-Aldrich, Gillingham, UK) and cultured with daily media changes.
2.2 Hydrostatic bioreactorCyclic hydrostatic pressures were applied to the 3D hydrogels and 2D cell cultures using a custom-made bioreactor designed and built in collaboration with Instron/Tissue Growth Technologies (Minnetonka, MN, USA) [1]. The bioreactor chamber is a sealed, anodised aluminium vessel accommodating a standard-sized cell culture plate (with the lid removed) allowing for pressure changes to be transferred to the gas phase above the well plate and transduced into the cell culture media. Compressed, recycled incubator air was fed from a continuously-running scroll compressor via a heater (to maintain the temperature of the inlet gasses at 37 °C) and through a system of valve gear into the chamber through a sterile cartridge filter. The gas was removed from the chamber by a vacuum mechanism and directed back into the incubator to be recycled. The operation of the valves and heater was controlled by TGT’s GrowthWorks software, allowing fine control of sinusoidal pressure waveforms from 0.0001 Hz to 2 Hz (or a constantly applied pressure) and between 0 and 280 kPa pressure.
2.3 Bioreactor stimulation regimesThe hydrogels were cultured for 28 days, with one hour per day in the hydrostatic bioreactor, the remainder of the experimental time period being under standard cell culture conditions in a conventional 37 °C 5% CO2 incubator. The pressure regimes applied were: A, No stimulation (static control in incubator); B, 280 kPa constant pressure with no cycling; C, 70 kPa at 1 Hz; D, 280 kPa at 0.05 Hz; E, 280 kPa at 1 Hz (Fig. 1). The hydrogels were cultured in 2 ml osteogenic media, which was removed and replaced daily.
Fig. 1
Hydrostatic pressure bioreactor: Experimental set up and loading regimes. A, Human MSCs were seeded in collagen hydrogels within transwell insert moulds suspended in a 24-well plate, B. 24 h after seeding, the plate containing the suspended hydrogels was placed into the anodised aluminium bioreactor chamber, C, which was sealed and connected to the bioreactor hardware, comprising a valve-based pressure regulator which used compressed, recirculated incubator air to apply pressure to the headspace above the culture plate at the required cycling regime, D. E shows representative traces of the loading regimes employed in this experiment: 0 kPa (static control); 280 kPa constant pressure; 0–70 kPa cycled at 1 Hz; 0–280 kPa cycled at 0.05 Hz; 0–280 kPa cycled at 1 Hz
2.4 X-ray microtomographyAnalysis of the cell-seeded hydrogels was by X-ray microtomography (μCT) using a Scanco μCT40 (beam energy: 55 kVp, beam intensity: 145 μA, 200 ms integration time, spatial resolution: 10 μm). The hydrogels were removed from culture at 7-day intervals and imaged under sterile conditions using X-ray microtomography (duration < 1 h) and returned to culture. The hydrogel scans were analysed at two density thresholds (50/1000 and 80/1000), firstly to determine the total size (volume) and average density of each hydrogel, and secondly at a higher threshold to determine the volume of the hydrogel that had become mineralised. These thresholds were determined from our previous research on foetal bone development and were conserved throughout these experiments, allowing for longitudinal comparison of hydrogel mineralisation across the investigation.
2.5 Analysis of ECM compositionFive of the six hydrogels from each group were assayed for ECM composition at day 28 of the experiment. The hydrogels were fixed in 4% paraformaldehyde for 20 min, then washed in PBS. The fixed hydrogels were imaged before and after the staining and destaining procedure using a stereoscopic dissecting microscope. Calcium deposition was determined by immersing the hydrogels in a 1% alizarin red solution for 4 h. The hydrogels were then immersed in several changes of water for 48 h to remove unbound dye, then imaged and individually destained in 2 ml 5% cetylpyridinium chloride solution, the absorbance of which was quantified using a spectrophotometer (at 562 nm) to determine the amount of calcium present in the hydrogels (all reagents from Sigma-Aldrich).
2.6 Polyacrylamide gels and surface functionalisationPolyacrylamide gel substrates were created with different stiffness and functionalised. 250 μl of 0.1 M sodium hydroxide (NaOH) was dispensed onto the surface of a 13 mm glass coverslip (Fisher Scientific, Loughborough, UK) and placed on a hotplate at 70 °C to allow evaporation and formation of an even coating. The coverslips were then coated with 200 μl (3-Aminopropyl)triethoxysilane (Sigma-Aldrich) for 5 min in a fume hood and washed for 30 s under running dH2O. The coverslips were then immersed in a 0.5% glutaraldehyde/PBS solution (Sigma-Aldrich) for 30 min and left to dry overnight. Chloro-silinated glass slides were prepared by coating glass slides in 100 μl dichlorodimethylsilane (Sigma-Aldrich) for 5 min in a fume hood, followed by rinsing for 30 s three times with dH2O. Solutions of Acrylamide/ Bisacrylamide (Sigma-Aldrich) were prepared in different ratios to generate substrates with stiffnesses of 1, 10 and 40 kPa, degassed in a vacuum desiccator for 15 min, and then mixed with 1/100 10% (w/v) ammonium persulfate (Sigma-Aldrich) by volume and 1/1000 N,N,N′,N′-Tetramethylethylenediamine (Sigma-Aldrich) by volume. 25 μl of the solution was pipetted onto prepared chloro-silinated slides and the amino-coated coverslips placed face down on top. The solutions were allowed to polymerise for 30 min, and then the polyacrylamide coated coverslips were removed from the cholo-silinated slides, transferred to well plates and washed twice in PBS. The coverslips in well plates were then sterilised overnight under UV light in a class II biological safety cabinet. The polyacrylamide substrates were then functionalised by immersion in 200 μg/ml sulfosuccinimidyl 6-(4'-azido-2'-nitrophenylamino) hexanoate (Sulpho-SANPAH) (Thermo-Fisher, Altrincham, UK) in DMSO/dH2O, cured under a 365 nm UV light source in a Bio-rad GenX UV chamber for 10 min, rinsed twice in 50 mM HEPES and 0.2 mg/ml Type I collagen solution added (BD Biosciences) and allowed to incubate overnight at 37 °C. The collagen solution was then aspirated and the coverslips washed first in PBS and then in culture media prior to cell seeding.
2.7 Immunofluorescence (actin and YAP)Monolayer cell cultures were washed briefly in PBS, fixed in 10% neutral-buffered formalin (Fisher Scientific) and permeabilised by adding 0.01% triton-X 100 (Sigma) for 15 min, followed by washing in PBS- 0.1% Tween (Sigma), and then blocking in 1% bovine serum albumin (BSA) (Fisher Scientific) for 1 h followed by two washes in PBS-0.1% Tween (all steps carried out at room temperature). Primary antibodies (anti-human YAP mouse monoclonal, Santa Cruz Bio, Dallas, TX, USA; anti-human osteocalcin mouse monoclonal, R&D systems Minneapolis, MN, USA; anti-human Runx2 goat monoclonal, R&D systems) were made to 2 μg/ml using a solution of 0.1% BSA in PBS with 0.1% v/v tween. The antibody solutions were incubated with the fixed cells overnight at 4 °C, then the cell layers washed twice in PBS-0.1% Tween and incubated with either Alexafloura 488 or 555 (2 μg/ml, 0.1% BSA, 0.1% Tween-20 in PBS) (Abcam, Waltham, MA, USA) in the dark for 1 h at room temperature, followed by two PBS-0.1% tween washes. Cell nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) for 30 min, and F-actin stained with Cytopainter 555 phalloidin (Abcam) for 1 h, with both steps performed at room temperature in the dark. The coverslips were mounted onto microscope slides with Vector shield medium (Vector Laboratories, Newark, CA, USA), and imaged on three fluorescence channels using an Olympus U-TBI 90 laser scanning confocal microscope. Images were acquired using the Fluoview10 software and analysed with Fiji/ImageJ software on a Win64 operating system (http://fiji.sc/Fiji).
2.8 Quantification of stainingFluorescence images were acquired with identical offset and gain settings for each sample and experiment, and fluorescence intensity analysed in ImageJ to quantify changes in the ratio of nuclear to cytoplasmic concentration of YAP. The nuclear fluorescence intensity values were subtracted from the total cell fluorescence values, and then normalised to the cell area to give a nuclear: cytoplasmic fluorescence intensity ratio per cell.
2.9 Statistical analysisNumerical data was analysed with GraphPad Prism 8 using a Student’s t-test or one-way analysis of variance (ANOVA) with multiple comparison tests. Significance was set at p < 0.05. Data are presented as mean values ± standard deviation.
留言 (0)