Adam19 has been consistently associated with pulmonary function in human GWAS. However, GWAS alone cannot establish causality. Mouse models are useful in investigating the causal role of loci identified in GWAS in pulmonary function. We successfully generated a novel Adam19 knockout mouse model and confirmed gene disruption through RNA-Seq and RT-qPCR analysis. Contrary to previous publications, our KO mice are viable and generally healthy, without the lethal cardiac abnormalities reported previously [19, 36].
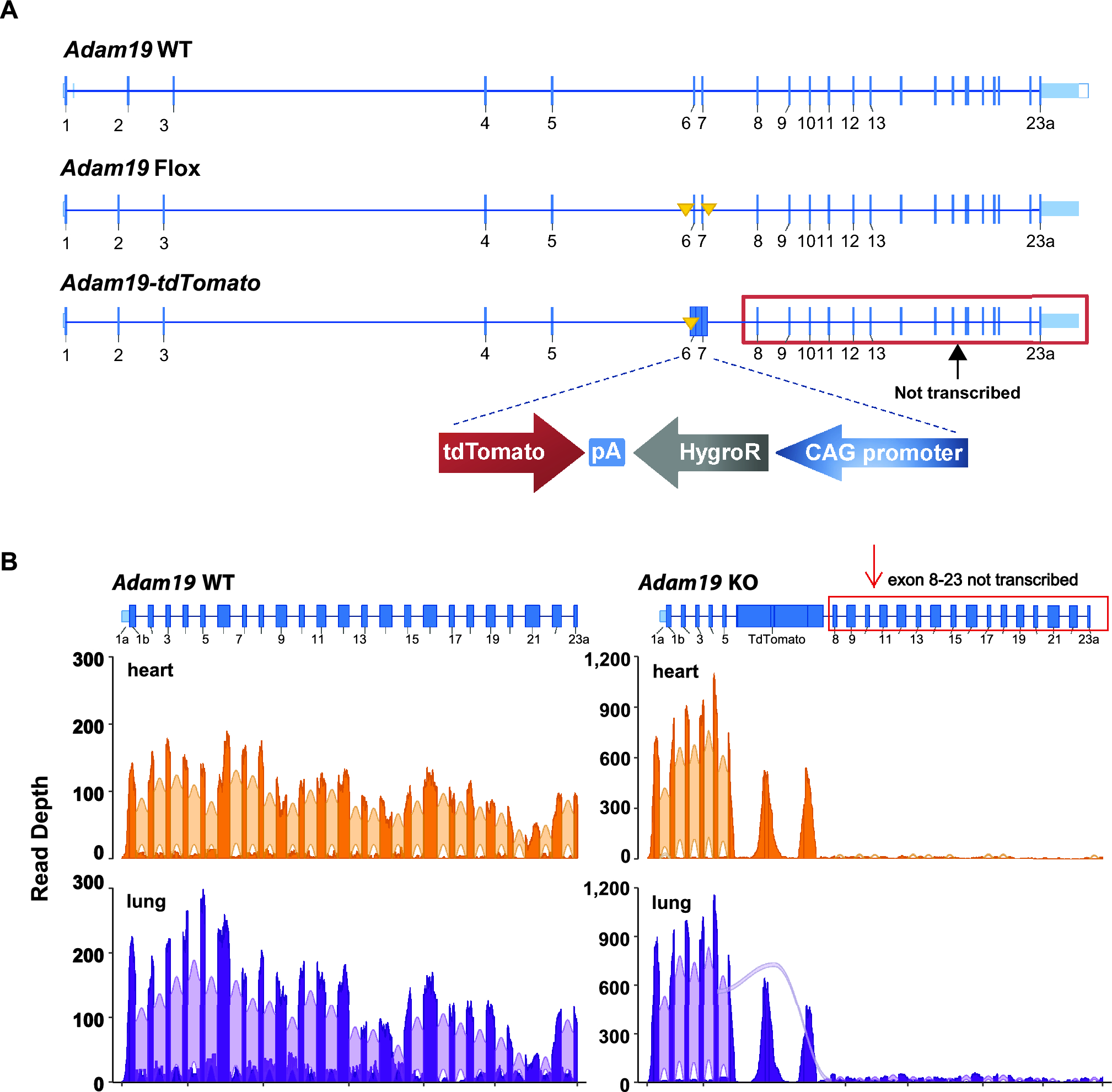
We considered factors potentially contributing to the discrepancy in the viability of our KO compared to prior work [19, 36]. Firstly, methods for producing the knockout differed between studies. Kurohara et al. [19] replaced exons 10 through 12 with an antisense Neomycin resistance cassette, and Zhou et al. [36] introduced a gene trap 3’ of exon 14. We replaced exons 6 and 7 with an in-frame tdTomato construct.
Second, genetic disruption of a multi-exon locus, like Adam19, may generate novel transcript variants through alternative splicing; some may result in a neomorph that rescues ADAM19 deficiency. In contrast, others may be potentially toxic in the absence of ADAM19. Our RNA-Seq analysis was designed to detect alternative splice variants but identified no alternative Adam19 splice variants and no active transcription from exons 8 through 23. Interestingly, we detected a novel splice variant in the lung, splicing from exon 5 to exon 8. However, this transcript led to a near-immediate nonsense mutation. Thirdly, the remaining gene structure in a knockout might influence its interaction with other genes or proteins, leading to different functional consequences. Our knockout mice expressed only the first five exons of Adam19, which do not include the sequences that encode the active catalytic sites of ADAM19. Transcriptions of exons 8 through 23 were nearly absent, providing confidence that no functional ADAM19 metalloproteinase domains were formed. While both genetic constructs in previous studies [19, 36] disrupt metalloprotease function, they may have generated truncated ADAM19 proteins that could interact with other proteins in a non-productive or dominant-negative manner.
Additional explanations for differences in the viability of the KO across studies include differences in the genetic background of the mouse lines used. Our Adam19-deficient allele was generated in 129S ES cells and subsequently maintained on the 129S6/SvEvTac background, whereas the other studies used mice with a mixed genetic background of 129 Sv and C57BL6/J [19, 36]. Further, in prior work, variability was seen in the penetrance of the observed cardiac phenotypes. In Kurohara’s ADAM19-deficient line, some mice survived to adulthood without severe cardiac defects besides enlarged hearts [19]. Partial penetrance of lethal phenotypes is common, so this phenotype variability is not surprising. However, it does suggest that the genetic disruption of Adam19 is more complex than initially envisioned during our gene targeting design.
We do not know why our Adam19-deficient mice were viable without the noticeable cardiac defects observed previously [19, 36]. However, all available evidence and data strongly indicate that we had a functional knockout of the canonical ADAM19 protein despite the small sample size as a limitation. Moreover, our knockout had normal-appearing hearts, and there were no obvious survival disadvantages. Our knockout mice unlikely retained ADAM19 activity, given that exons 1 to 5 only encode for the first 111 of 920 amino acids of canonical ADAM19 protein but none of the active sites of metalloproteinase domains. In addition, Adam19’s first five exons appeared to have higher expression in heart and lung samples in KO than wildtype, possibly caused by either a feedback mechanism attempting to compensate for the functional loss of Adam19 or by the absence of appropriate 3′ UTR elements for the consistent transcript turnover in mutant samples or both.
Our Adam19 KO animals exhibited several notable phenotypic differences compared to their WT littermates, including reduced body weight, decreased tibia length, and altered body composition. Inoue et al. reported that Adam19 was involved in osteoblast differentiation in mice [38], which may help explain why our Adam19 knockouts have shorter tibias. Weerasekera et al. demonstrated a correlation between high ADAM19 expression in human peripheral blood mononuclear cells and BMI, relative fat, and TNF levels [37]. They also observed increased Adam19 mRNA and ADAM19 protein in the liver tissue of mice fed a high-fat diet (HFD). In contrast, neutralizing ADAM19 protein with its antibody resulted in weight loss, reduced white fat accumulation, and decreased TNF protein levels in the liver of HFD-fed mice. These published findings provide insights into our observations of smaller body sizes, reduced body weight, and altered body compositions. Further, differentially expressed genes we identified included Kpna2, which was associated with body weight and BMI in human GWAS [39], and Cd300lg, which has been associated with increased intramyocellular lipid content and reduced fasting forearm glucose uptake in humans [40]. Additionally, GSEA enrichment in multiple pathways related to cell proliferation and metabolism could contribute to the anthropometric phenotype in our KO. Collectively, our data support the role of ADAM19 in regulating growth and body weight development.
Human GWAS have identified hundreds of variants in or near ADAM19 that are significantly associated with lung function [2, 3, 5, 7, 10, 41]. In particular, the minor alleles of sentinel SNPs have been associated with lower FEV1/FVC and FEV1, including rs2277027 [5], rs11134789 [3, 10], and rs4331881 [2], which are in high linkage disequilibrium. However, other genome-wide significant variants displayed opposite effects with the minor allele associated with higher FEV1/FVC and FEV1, including rs1990950 and rs59327154. Several variants in ADAM19 have also been associated with COPD, including rs2277027, rs1422795, rs11744671, and rs113897301, for which the minor allele was associated with an increased risk of COPD [9, 42, 43]. When we queried the Genotype-Tissue Expression (GTEx) Portal for gene expression, we noted hundreds of variants in ADAM19 that implicate significant eQTLs in lung tissue, including sentinel SNPs rs11134789 and rs2277027, which were among the top most significant eQTLs and had minor alleles associated with increased expression [44]. Yet, other variants that had significant positive associations in GWAS had minor alleles associated with decreased expression. In a study combining UK Biobank GWAS data with gene expression data, protein level data and functional annotation, ADAM19 met the criteria as a putative causal gene for FEV1/FVC as well as FEV1 and peak expiratory flow [3]. However, given the large number of ADAM19 variants associated with lung function and COPD, as well as gene expression and the range of effects depending on the individual SNPs, it is difficult to pinpoint a single causal variant and, therefore, challenging to comment with certainty on the overall direction of effect. This is a known limitation of GWAS and highlights the importance of follow-up research utilizing fine-mapping and multi-omics data [45, 46] as well as mouse model approaches.
Critical to comparison with human GWAS, the Adam19 KO mice also displayed altered baseline pulmonary function parameters, namely decreased elastance of the respiratory system, minute of work of breathing, tissue damping, tissue elastance, and declined forced expiratory flow at 50% forced vital capacity, as well as increased FEV0.1 and FVC. Because of the smaller size of our KO, we adjusted all statistical analyses of lung function parameters for weight to ensure the observed lung function differences by genotype were not due to the smaller size. Using flexiVent, lung function parameters were determined based on lung responses to frequency-dependent input signals. For input signals at a fixed breathing frequency, lung function parameters (Rrs, Ers) were captured. The Rrs and Ers reflect resistance and distensibility of the whole respiratory system, including airways, lung tissues, and chest wall [47]. For input signals at various frequencies, lung function parameters can be partitioned to reflect the contribution of different lung regions [48]. For example, Rn reflects the resistance of central airways. Tissue damping (G) closely relates to the resistance of peripheral lung tissue. Tissue elastance (H) reflects the elastic recoil of lung tissue. FEV0.1 simulates human FEV1 and FEV0.1/FVC simulates human FEV1/FVC. FVC is relevant to FVC in humans. In our knockout, we found consistent results for FEV0.1 and FVC. Perhaps, not surprisingly, we did not find a significant difference when taking the ratio. FEV0.1 was higher, while FEF50 was lower in our KO compared to WT; we would not necessarily expect directions of effect to be the same because the two parameters were uncorrelated in our data. We note that Kwon et al. reported mild COPD patients with normal FEV1 had reduced FEF25%-75%, which is equivalent to FEF50 in our study [49]. mWOB is correlated with airway compliance and was reduced in an emphysema mouse model and increased in a fibrosis mouse model [50]. Our data provide compelling evidence for a causal role of ADAM19 in pulmonary function, confirming findings from human GWAS.
Collagen is the main constituent of lung connective tissues, which provides support in the bronchi, interstitium, and alveolar wall structures and plays an essential role in lung mechanics [51]. We did not observe any genotype differences between WT and KO for either lung histology (Fig. S5) in general or for collagen deposition (Fig. S9). This is consistent with our lung function findings—no change in the naïve KO for the baseline respiratory system resistance (Rrs) and Newtonian resistance (RN, reflecting conducting airway resistance) (Fig. 4) and the airway responsiveness to methacholine (Fig. S7). Of note, lung function differences were generally subtle, and thus, the lack of histologic differences is perhaps not surprising. As a limitation, our study did not investigate the roles of other connective tissue components in lung function.
The precise molecular mechanisms underlying these observations for lung function remain unknown. ADAM19 cleaves NEUREGULIN-1 (NRG1), an erythroblastic leukemia viral oncogene homolog (ERBB) receptor tyrosine kinases ligand. ERBB receptor ligands NRG1 and epidermal growth factor affect fetal surfactant synthesis in the developing mouse lungs [52]. ADAM19 has also been implicated in non-proteolytic functions, such as regulating neuromuscular junctions in murine embryos through Eph family receptor-interacting proteins (EPHRIN)-A5/EPHRIN-A4 signaling [53]. In addition, the cytoplasmic tail of ADAM19 has several Src homology 3 (SH3) binding sites that regulate protein–protein interactions. ADAM19 binds strongly to the scaffolding protein tyrosine kinase substrate with five SH3 domains and the Src tyrosine kinase, potentially influencing cytoskeletal functions that impact cell motility, contractility or tissue development [54]. Therefore, disruption of ADAM19 may have important effects on lung development, neuromuscular functions, tissue elastance, contractility, or other unidentified signaling processes.
Our differential gene expression analysis identified genes related to lung physiology and pathology. For example, increased Kpna2 expression may contribute to altered lung function, consistent with publications that KPNA2 genetic variation is associated with FEV1/FVC in human GWAS [2] and plays a role in lung cancer [55]. Our KO had increased Pttg1 expression. Pttg1 is involved in cell cycle regulation [56] and the development of lung cancer [57], suggesting its role in the lungs. Interestingly, some of these differentially expressed genes were on chromosome 11, where Adam19 is located; this might imply additional effects of the KO construct. Chromosome 11 has a high gene density; the genes we detected on chromosome 11 were proportional to the number of genes on other chromosomes. The gene expression differences were relatively small between our Adam19 WT and KO. We confirmed 1) the absence of Adam19 transcription and 2) the absence of novel Adam19 splicing variants that might rescue the lethal cardiac phenotype as expected based on the literature, which were our primary goals of the RNA-Seq analyses. The small sample size was a limitation to identify the differentially expressed genes across the genome definitively. We identified a limited set of differentially expressed genes with this modest sample size.
Given our observation of reduced neutrophil infiltration in BALF following LPS exposure in the Adam19 knockout, we investigated whether airway responsiveness to methacholine differs between KO and WT mice following LPS administration. Notably, our knockout mice showed decreased tissue damping and tissue elastance response to methacholine following LPS exposure compared to WT, indicating an attenuated response to inflammation. ADAM19 facilitates the release of TNF from the cell membrane, promoting an inflammatory response and contributing to the development of inflammatory diseases [17, 18, 37, 58]. We did not identify genotype differences of cytokine changes following LPS (vs. saline). This could reflect the limited number of cytokines examined, which is a limitation of our study. However, GSEA identified the enrichment of downregulated differentially expressed genes in TNF signaling pathways in our Adam19 KO mice. This is consistent with these previous findings [17, 18, 37, 58] and helps explain the reduced lung functional response to the inflammation in our knockout mice.
In summary, we created a viable whole-body Adam19 knockout and used this model to examine the role of Adam19 in lung function, following up on findings from human GWAS implicating this gene. In addition to smaller body size, the lack of functional Adam19 resulted in reduced respiratory system elastance, minute work of breathing, tissue elastance, forced expiratory flow at 50% FVC, and increased FEV0.1 and FVC. Pathway analysis of genes differentially expressed after disruption of Adam19 implicates pathways crucial in lung inflammation, including TNF signaling pathways. Our data provide evidence to support a causal role for Adam19 in regulating pulmonary function development. Although our study is limited to a descriptive scope and a definitive understanding of mechanisms underlying our findings requires further investigation, our novel Adam19 KO murine model could be helpful in future studies to dissect the role of this gene in lung function.

留言 (0)