
記住我
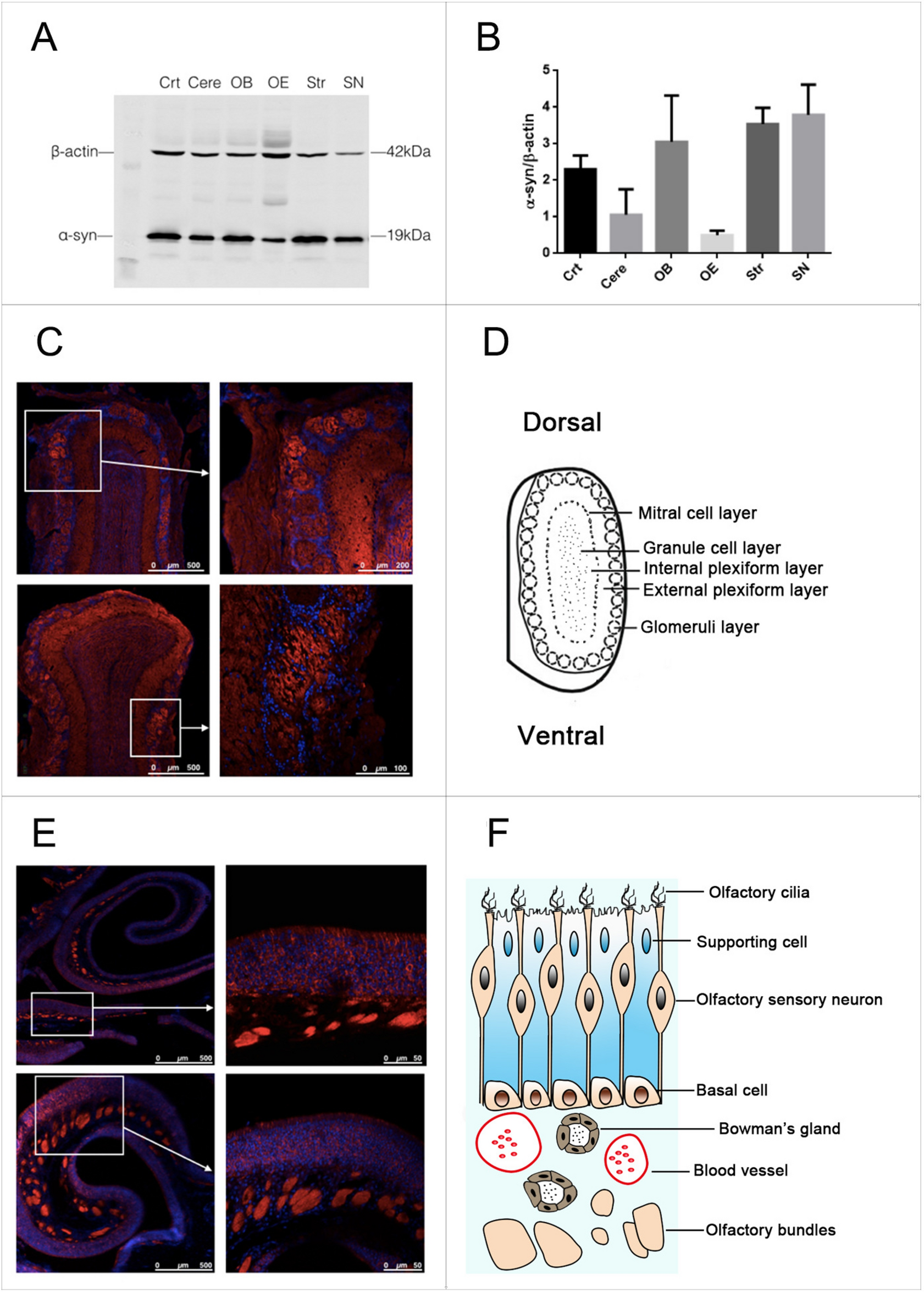


A systematic literature review was conducted utilizing PubMed as the primary database. The review honed in on articles originally published in English from a range of nations, including the United States, United Kingdom, Germany, Australia, Japan, South Korea, and China. These countries were strategically chosen due to their active research communities dedicated to IRDs, notably STGD. In order to focus on the latest and most innovative therapeutic approaches, literature pertaining to these approaches was systematically limited to works with a publication date within the last ten years. An overview of the systematic literature review process, including the focused search categories and the specific keywords used was compiled into a table (Table 1). Details from each clinical trial were retrieved from ClinicalTrials.gov (US).
Table 1 Comprehensive literature search strategy for STGD therapies and diagnosticsAfter these preliminary searches, the list of articles was further narrowed by only including publications that emphasized one of the following criteria: therapeutic innovation, clinical trial relevance, clinical applicability, ABCA4-associated retinopathies, and/or human genome. Given the extensive body of literature in this field, it was challenging to conduct a thorough review of the literature. The chosen works for this review are deemed representative of the latest advancements in precision medicine therapies for STGD.
Drug therapies for STGDDrug therapies aim to address the underlying cellular dysfunction, such as the accumulation of toxic substances like lipofuscin in the retina or the degeneration of photoreceptor cells. These types of therapies are advantageous because they have the potential to stabilize or slow the progression of the disease through systemic treatment. Researchers are investigating various compounds that can reduce visual cell damage, manage oxidative stress, or improve the metabolic health of retinal cells. Currently, there are numerous clinical trials that explore compound therapies that target the critical pathological processes associated with STGD [26]. As of the current “state-of-the-science,” there are no drug therapies specifically approved for STGD. Treatment mainly focuses on managing symptoms and preventing further deterioration. Patients are advised to avoid high-light environments and use protective lenses to shield their eyes from potential damage. Behavioral modifications, such as using low vision aids and making lifestyle adjustments, are recommended in order to enhance daily functioning and preserve the remaining vision. Research continues into pharmacological treatments, but as of now, the focus remains on supportive care and mitigating the impact of the disease on patients’ lives.
The link between lipofuscin accumulation and complement system activation in RPE cells, a layer of cells behind the retina that nourish photoreceptor cells, as observed in the ABCA4 knockout model, extends beyond cell lines to more complex organisms [27]. Since complement activation plays a significant role in the development of AMD, the similar characteristics shared between this condition and STGD suggest that medications developed for AMD could also be applicable to STGD [28]. The translational research, bridging the gap from cellular studies to more advanced models, often employs animal models such as mice genetically modified to mimic STGD, and in some cases, primate or porcine models, to replicate the disease’s progression and test potential treatments. Avacincaptad pegol (Zimura), a complement inhibitor, exemplifies this translational journey. Initially tested in cell lines and animal models, Zimura’s effectiveness in inhibiting the C5 component of the complement system and its downstream effects, C5a and C5b, has been established. Its intravitreous injection reduces chronic inflammation and cell death in the macula, thereby slowing the degeneration of RPE cells [29]. This pathway from basic research to clinical application demonstrates the interconnectedness of various research methodologies in developing treatments for complex diseases like STGD.
An ongoing Phase 2b clinical trial is currently underway to evaluate the safety and effectiveness of a Zimura intravitreal injection compared to a Sham (placebo-like) injection in patients with STGD. Patients of all sexes aged 18–60 are eligible for this study, and their best-corrected visual acuity (BCVA) must be between 20/20 and 20/200 (National Library of Medicine [NLM], NCT03364153). Approximately 120 people from around the world are enrolled in the study in 35 centers, and each participant will receive 36 injections. The study will continue for approximately 18 months; results have not yet been reported. A Phase II/III trial in 286 patients with AMD found that Zimura was well-tolerated and effective (NLM, NCT02686658). The participants will be randomly assigned to one of the following groups: Zimura and Sham (an empty syringe with no needle to prevent bias in results).
Vitamin A (all-trans-retinol) is a common target of the visual cycle and is a precursor to 11-cis-retinol. It is presumed that decreasing vitamin A levels may inhibit the synthesis of all-trans-retinol and, consequently, lipofuscin [26]. Although conventional vitamin A supplements could be harmful, as seen in the ABCA4 knockout model involving genetically modified mice where the ABCA4 gene is inactivated [27], an alternative strategy involves administering deuterated vitamin A (ALK-001). ALK-001 (gildeuretinol) is an altered form of vitamin A that slowly forms vitamin A dimers in the body by replacing its natural form [30]. It is the first medicine that is being clinically developed to treat STGD. Compared to traditional vitamin A supplementation, ALK-001 can minimize the formation of toxic vitamin A dimers that contribute to retinal degradation in STGD. This occurs by modifying the structure of the vitamin A molecule so that three hydrogen atoms are replaced with deuterium atoms. ALK-001’s modified structure allows it to specifically target and deliver vitamin A to the retina [30]. Researchers have found that ALK-001 slowed the rate of dimerization of vitamin A by 4–5 times in in vitro studies and by 80% in in vivo mouse models, which leads to the preservation of visual function in animal models of STGD [31]. An active clinical trial is currently in Phase 2 to evaluate how orally administered ALK-001 affects the progression of ABCA4-related STGD. Participants in the study can be male or female and must be between 8 and 70 years old with any VA. According to the results, the growth rate of atrophic lesions in the ALK-001-treated group was 21% slower than that in the untreated group (NLM, NCT02402660). Furthermore, the results of the study have shown that this is the first instance where a therapeutic intervention has demonstrated a clinically and statistically significant slowdown in the progression of STGD.
Building on the theme of targeting vitamin A-related pathways in STGD, other therapeutic approaches are being explored. Tinlarebant (LBS-008) is one such oral drug therapy. It operates by inhibiting the activity of RBP4 (retinol-binding protein 4), which is responsible for transporting retinol to the eye [32]. By reducing the transport of retinol, Tinlarebant aims to decrease the accumulation of harmful substances in the retina, thereby lowering the risk of blindness associated with STGD. An ongoing clinical trial currently in Phase 3 is assessing how effective Tinlarebant is in slowing the progression of atrophic lesions in adolescents with STGD. Participants will take 5 mg of Tinlarebant/placebo for up to 24 months (NLM, NCT05244304).
Additionally, metformin hydrochloride, commonly prescribed for the treatment of type 2 diabetes, is being investigated for its potential benefits in retinal degeneration. A correlation between metformin use and the slowing of the progression of retinal degeneration has been discovered because metformin increases macroautophagy through the mammalian target of the rapamycin complex 1/AMP-activated kinase pathway [33]. The stimulation of this pathway allows the RPE to better handle lipofuscin. This approach is being tested in a Phase 2 clinical trial to determine its safety and efficacy in slowing the rate of change in the degeneration of photoreceptors in ABCA4 retinopathy (NLM, NCT04545736).
The realm of drug therapies for STGD is witnessing a significant evolution, with several promising strategies currently being investigated. From the use of modified vitamin A derivatives such as ALK-001 to innovative approaches such as Tinlarebant that inhibits the transport of retinol to the eye and the repurposing of metformin to slow retinal degeneration, the landscape of treatment options is expanding. These therapies, each with their unique mechanisms of action, are at various stages of clinical trials and offer hope for more effective management of STGD. The results of these studies could not only revolutionize the treatment of this specific retinal disease but also provide valuable insights into managing other similar retinal disorders, paving the way for a future where vision loss due to genetic conditions can be significantly mitigated or even prevented.
Gene therapies for STGDSince a significant portion (90–95%) of STGD cases stem from loss-of-function mutations in the ABCA4 gene and are inherited in an autosomal recessive manner, gene therapy presents a logical strategy to both prevent and slow the progression of retinopathy associated with ABCA4 [34]. Every year, there are new ongoing clinical trials for gene therapy treatments for IRDs in which a pathogenic gene is replaced with a functional gene [35]. Furthermore, delivering large DNA molecules, which is required for treating Stargardt disease, remains a challenge. This is due to the preferred vector being adeno-associated virus (AAV), a small, single-stranded DNA dependovirus. This virus belongs to the parvovirus family and has a packaging capacity of ~ 4.7 kb [36]. Therefore, strategies to facilitate large gene delivery, such as dual-AAV, lentiviral, and nonviral platforms, are being developed.
Lentiviral vectorsSince the human ABCA4 cDNA is almost 7 kb, lentiviral vectors are an appropriate choice for gene therapy because they have a packaging capacity of ~ 8 kb and carry large expression cassettes [37]. They deliver therapeutic genes into the host genome of transduced cells in vivo and transduce nondividing cells, which is necessary for terminally differentiated cells such as photoreceptors [38]. These vectors have the potential to halt or reverse the accumulation of lipofuscin, preserving photoreceptor function over time. The targeted delivery of lentiviral vectors to the retina can be achieved through subretinal injections, a method that has been refined to increase precision and reduce invasiveness. Furthermore, the development of lentiviral vectors that express ABCA4 under the control of retina-specific promoters can enhance the specificity of expression, reducing the possibility of off-target effects and maximizing therapeutic benefits.
In the field of eye diseases, the unique aspect of using one eye as a control in clinical trials is particularly intriguing. This approach was evident in the clinical application of a novel gene therapy technology, where a subretinal injection of EIAV-ABCA4 (equine infectious anemia virus) was administered to the worse-seeing eye of 22 patients. They were followed for three years to assess the safety and efficacy of this treatment. The use of the worse-seeing eye as the treatment site allows for direct comparison with the patient’s other eye, serving as a natural control. SAR422459, the drug used in this study sponsored by Sanofi (NLM, NCT01367444), exemplifies the lentiviral vector’s potential. It utilizes a self-inactivating EIAV vector designed to deliver the ABCA4 gene directly to the affected retinal cells. While 27% of treated eyes experienced worsening of retinal pigment epithelium atrophy as observed on fundus autofluorescence, an imaging technique that displays retinal changes, there was also a noteworthy positive outcome. One treated eye from the cohort receiving the highest dose experienced a reduction in macular flecks. This study was discontinued following a reassessment of clinical development objectives. However, the initial safety outcomes observed over the first year were encouraging. Participants reported 125 adverse events, all of which were classified as mild to moderate in severity [39]. A second long-term dose-escalation phase I/II trial explores subretinal injection of an ABCA4-carrying lentivirus in 27 patients (NLM, NCT01736592).
Adeno-associated viral vectorsIn parallel, the use of adeno-associated viral (AAV) vectors has also gained prominence in gene therapy research for retinal diseases. Over the past two decades, AAV vectors have been a focal point due to their safety profile and efficiency in gene delivery [40]. These vectors are being extensively studied for a variety of IRDSs, including STGD, offering another avenue for therapeutic intervention. The exploration of different viral vectors - lentiviral and AAV - reflects the dynamic nature of gene therapy research in ophthalmology. Each vector type has unique characteristics and potential applications, contributing to the broader goal of developing effective gene therapies.
AAVs have emerged as the preferred vector for gene therapy due to their favorable safety profile with few adverse effects [35]. However, a significant hurdle persists: the limited capacity of AAV vectors to carry only small genes, which leaves many monogenic diseases untreatable using existing technologies. Initial techniques focused on engineering a single large transgene with the complete 6.8 kb sequence of the ABCA4 gene, and the first AAV vector-based gene therapy product for an IRD was approved in December 2017. However, the addition of necessary regulatory sequences increased the size to over 10 kb, beyond the typical packaging limit of AAV vectors. Despite this, researchers managed to encapsulate and express the full-length ABCA4 protein in vitro and in vivo [41]. The AAV packaging process, however, is unregulated and results in varied incomplete transgene versions within the vector shells, known as a fragmented dual-vector approach [42]. The effectiveness of this method depended on serendipitous overlaps between the truncated transgenes [43]. Further assessments showed that the fragmented AAV-ABCA4 vectors could recombine to create the full ABCA4 gene transcript but generated hybrid transcripts with unintentional AAV genome integrations [44]. Although initial attempts to package an oversized AAV transgene with the complete ABCA4 were successful, it became clear that this strategy was not reasonable because it is challenging to produce homogeneous AAV preparations containing the intended transgene. Hence, the focus shifted to dual AAV vector strategies in light of the potential therapeutic advantages of delivering a functional ABCA4 gene.
The advancements in using AAV vectors for gene therapy, particularly for conditions like STGD, have originated from a combination of laboratory research using human cell lines and in vivo studies in animal models. In labs, researchers often use human retinal cell lines, including RPE cells, to study the behavior of AAV vectors and the expression of the ABCA4 gene. These cell lines are important for understanding how the therapy might work in human tissues and for testing the safety and efficacy of different vector designs. Mouse models play a significant role in gene therapy research. Mice genetically modified to mimic the genetic and physiological aspects of STGD provide a platform for testing AAV vectors in a living organism. These models allow researchers to observe how the therapy affects the retina over time and to identify potential side effects or complications. Initial strategies focused on engineering a single large transgene with the complete sequence of the ABCA4 gene. The challenge of including necessary regulatory sequences and the limitations of AAV vectors’ capacity led to the development of fragmented and dual-vector approaches.
Dual AAV vectors are a method used in gene therapy when the gene fixed into a patient’s cells is too big for one virus to carry. If the gene cannot fit into a single AAV vector, scientists split it into halves (< 5 kb in size) [45]. One vector contains the promoter and the 5’ portion of the cDNA, while the other contains the polyA signal and the 3’ portion [46]. Once inside the target retinal cells, these split gene segments can reassemble into the complete, functioning gene. This reassembled ABCA4 gene can produce the correct protein, which is vital for the visual cycle and prevents the accumulation of lipofuscin and bisretinoid. The results of a study in mouse models demonstrate that delivering the ABCA4 protein via dual AAV vectors significantly reduces lipofuscin in the ABCA4 animal model of STGD [46].
AAVantgarde Bio, an Italian headquartered biotechnology company, is working to validate an AAV-based large gene delivery platform named AAV intein. Inteins carry out a process known as protein splicing, a multi-step biochemical reaction made up of the cleavage and formation of peptide bonds [47]. AAV intein vectors mediate the expression of large therapeutic proteins in the retina and in human cell lines derived from RPE cells. In STGD mouse models, subretinal administration of AAV intein vectors improved the retinal phenotype, which demonstrated the potential of AAV intein for gene therapy of these types of blinding diseases [48]. Preclinical studies are currently ongoing for the AAV intein platform. Preliminary results presented at the 2024 Association for Research in Vision and Ophthalmology (ARVO) showed positive non-human primate (NHP) safety and efficacy data at demonstrating that the intein vectors can be administered safely to NHPs. This confirms that the AAV intein-mediated retinal gene therapy for STGD is safe and effective in relevant animal models, moving closer towards the clinical translation of this platform.
Similarly, other researchers are developing innovative strategies to overcome the limitations of using a single AAV vector. The American biotechnology company Applied Genetics Technologies Corporation (AGTC) also announced a dual vector gene therapy system in 2019 developed by researchers from the Department of Ophthalmology at the University of Florida designed to accommodate larger genes within the retina, such as ABCA4. The two DNA fragments recombine once inside the cell and reconstruct the entirety of the ABCA4 coding sequence. The researchers found that after subretinal injection of dual AAV into KO mice lacking the ABCA4 gene, the full-length ABCA4 protein correctly localized to photoreceptor outer segments. These outer segments are key structures in photoreceptor cells because they convert light into visual signals and are the primary site where the ABCA4 protein functions in the visual process [7]. Furthermore, there was a reduction in the accumulation of lipofuscin. ATGC is moving forward with the preclinical development of the therapy, having shared data that demonstrates that it is effective in non-human primates [46]. William Hauswirth, the leader of the research team at UF, has pointed out the significance of AGTC’s success in safely delivering and expressing the ABCA4 protein in these primates through subretinal injection using their optimized vectors.
In a similar pursuit of innovation, ViGeneron has three novel next-generation gene therapy platforms that aim to target the drawbacks of current AAV therapies: vgAAV vector platform, REVeRT technology platform, and AAV Transactivation. The vgAAV vector platform enhances the transduction efficiency of target cells, facilitating minimally invasive administration methods such as intravitreal injections (Fig. 3). The second platform is REVeRT (REconstitution Via mRNA Trans-splicing) technology, which enables large genes (> 5 kb) to be effectively reconstituted in tissues, including the heart, brain, liver, skeletal muscle, and retina. In a mouse model of STGD, the researchers demonstrated high expression of ABCA4. Preliminary experimental data has suggested a potential improvement in retinal function. Finally, AAV transactivation utilizes CRISPR‒Cas technology to allow for the in vivo regulation of one or more genes. Its program, VG801, focuses on transferring the ABCA4 gene in a mouse model for STGD. The gene was transferred using REVeRT technology to account for the large size of ABCA4.
Nonviral vectorsAdditionally, despite the success of viral gene delivery in clinical trials, issues related to host immune responses and oncogenesis have been reported. As the industry continues to explore the full potential of AAV vectors, attention is also shifting toward the promising realm of nonviral delivery systems. These systems are particularly compelling due to their ability to overcome some of the inherent limitations of viral vectors, such as the restriction on the size of the genetic payload and concerns about immune responses. Intergalactic Therapeutics uses synthetic biology and engineered gene circuits to deliver vectors in a nonviral way by making covalently closed and circular DNA molecules (C3DNA). C3DNA enables DNA vectors that are too large for viral delivery systems to be delivered without being inserted into the host genome, thus reducing the risk of insertional mutagenesis [49]. The data from its lead program, IG-002, demonstrated that subretinal administration of a DNA payload that encodes the human ABCA4 gene expressed human ABCA4 protein in adult porcine retinas over the course of 12 months. The C3DNA system, in combination with COMET delivery, shows great potential for gene therapy, especially for diseases requiring large DNA payloads. Intergalactic has plans to move its program into the clinic in 2024. This advancement could broaden the scope of treatable genetic conditions, signaling a new chapter in gene therapy that complements the ongoing successes of viral vector-based approaches such as those pursued by Ocugen.
Base editingThe evolving landscape of gene therapy paves the way for advanced genome editing techniques such as base editing. Base editing is different from other gene editing tools, such as CRISPR-Cas systems that typically create double-stranded breaks in DNA. Base editing offers a more targeted approach than traditional gene therapies, allowing for the direct, irreversible conversion of one DNA base into another, effectively correcting point mutations without making double-stranded DNA breaks (Fig. 4) [50]. This process is similar to fixing a single typo in a text. CRISPR-Cas-based editing systems have also been adapted for base editing purposes. These adaptations involve coupling the CRISPR machinery with enzymes that can modify DNA bases directly, thus enabling precise alterations at the genetic level without the need for breaking and repairing the DNA strands [51].
The Institute of Molecular and Clinical Ophthalmology Basel is working with Beam Therapeutics in Boston, Massachusetts to bring this innovative technology to the clinic. The goal of their project is to transfer the specific base-editor complex to patients’ retinas in the hopes of correcting the G1961E mutation. Base editing works by introducing single-nucleotide variants into DNA or RNA in living cells [52]. Mutations in the G1961E gene lead to the death of photoreceptors and RPE, and the demonstration of precise base correction in a high percentage of target cells is an essential requirement for the therapeutic application of base editing. Adenine base editing is a rational therapy for correcting the most common mutation in STGD because, at the optimal dose level, the optimized gene therapy vector in the study led to 40% editing rates in cones, 28% in rods, and 73% in RPE cells [53].
Overall, base editing is a highly promising development in the field of gene therapy, but whether it is more promising than the therapies previously mentioned in this review depends on various factors, including the mutation type and the maturity of the technology. Even though there are reports demonstrating the effectiveness of base editing in cell lines and mice, high levels of gene correction have not yet been demonstrated in human tissues and non-human primates [53]. Furthermore, the applicability of base editing is currently limited to certain types of mutations, such as transition mutations. Other therapies being developed, such as those utilizing AAV vectors or nonviral delivery methods like C3DNA, may be more suitable for a broader range of genetic alterations. However, whether it is more promising than the other gene therapies in this review cannot be fully determined until more data from comparative clinical trials becomes available.
OptogeneticsOptogenetics is a groundbreaking technique that merges genetic engineering with light to control the activity of individual neurons within living tissue [54]. This method involves the introduction of light-sensitive proteins, known as opsins, which are naturally occurring in certain algae and microorganisms, into specific types of cells, such as neurons in the retina [55]. Opsins are membrane proteins found in the photoreceptor cells of the retina (Fig. 5). Light interacts with opsins when it enters the eye and reaches the retina, which generates electrical signals that are transmitted to the brain by the optic nerve to be interpreted as visual information [56]. Nanoscope Therapeutics Inc. has come out with a novel therapeutic, Multi-Characteristic Opsin (vMCO-010), an ambient light activatable optogenetic therapy that works to restore vision in the advanced stages of STGD by restoring broadband light sensitivity, the ability of these introduced opsins to respond to a wide range of light wavelengths [56]. This broad sensitivity allows treated retinal cells, which are not primarily light-sensitive, to capture a wider spectrum of light, mimicking the function of healthy photoreceptors and generating neural signals for visual perception. A Phase 2 clinical trial completed in September 2023 evaluated the safety and efficacy of a single intravitreal injection of MCO-010 in 6 subjects with STGD (NLM, NCT05417126). The results were promising; patients demonstrated clinically meaningful improvements in BCVA, and there was a ~ 3 dB gain in mean light sensitivity measured by Octopus visual field perimetry.
Cell replacement therapy for STGDAs the degeneration of RPE and photoreceptor cells leads to diminished VA in the advanced stages of STGD, the treatment approaches discussed so far primarily aim to halt further vision loss. In order to restore vision that has already been lost, transplanting new cells is necessary. Encouraging proof-of-principle data has emerged from pre-clinical studies that involve introducing human embryonic stem cell (hESC)-derived RPE cells into the subretinal space of mice [57]. Regenerative medicine holds immense potential benefits for patients suffering from blinding eye diseases. Due to their unlimited proliferation and ability to differentiate into various cell types, stem cells are a valuable resource for tissue transplantation, and this type of therapy has the potential to treat STGD by regenerating the RPE. The stem cells are classified based on their potency, which refers to the range of cell types they can differentiate into and their origin [58]. The use of stem cells in therapy encompasses various approaches, with cell replacement therapy being the most recognized. In this method, stem cells are transformed into the specific cell type needed and then transplanted into the damaged tissue, where they integrate and work to restore its function [59].
Embryonic stem cellsEmbryonic stem cells (ESCs) are a source of therapeutic cells that can treat diseases due to dysfunction or tissue loss. Using this technology to treat diseases that impact the eye is an appealing first-in-human application. Genetically altered photoreceptor outer segments are often the cause of RPE degeneration in STGD and in preclinical macular degeneration models, which often utilize genetically modified animal subjects or cell cultures to replicate aspects of the disease, there is proof that subretinal transplantation of hESC-derived retinal pigment epithelium can save photoreceptors and stop vision loss [60]. The transplanted hESC-derived RPE cells provided the necessary support and functionality that the damaged native RPE cells could no longer offer. As a result, this intervention was shown to preserve photoreceptor cells and halt or slow down the progression of vision loss in these models. This study provided essential insights into the possibility of using stem cell-derived RPE cells for treating retinal diseases. It laid the groundwork for future studies and clinical trials and offered a new way to restore vision in diseases characterized by RPE degeneration. Following the development of the first hESC line by James Thompson and colleagues (1998), the application of hESC lines and their derivatives in both non-therapeutic and therapeutic contexts has ushered in a new era of innovation in modern medicine.
hESC-derived cell therapies have undergone intense scrutiny regarding ethical, regulatory, and biological safety aspects for human applications. After the Geron Corporation halted its pioneering hESC-based therapy for spinal cord injury in 2011, the Astellas Institute of Regenerative Medicine took the lead. They initiated the first hESC-derived RPE clinical trials for retinal degenerative diseases such as STGD and Geographic Atrophy (GA) due to AMD. Beginning in April 2011 and concluding in 2017, three Phase 1/2 clinical trials tested the MA09-hRPE cell line in suspension form across major eye centers in the United States, United Kingdom, and South Korea. These trials (UK-SMD: NCT01469832; US-SMD: NCT01345006; US-AMD: NCT01344993) have offered preliminary insights into the potential therapeutic benefits and behavior of the MA09-hRPE cell line in severely damaged subretinal environments in advanced STGD cases. Many patients in these trials had clinically measurable visual improvements [61]. By replacing the RPE cells lost in diseased retinas, hRPE transplants ultimately aim to restore the anatomical and functional integrity of the blood-retinal barrier (BRB) [62]. This restoration is crucial as the BRB is responsible for regulating ion, protein, and water flux into and out of the retina [63].
Advancements in multimodal imaging and functional testing, including adaptive optics-based scanning laser ophthalmoscopy and microperimetry, will be crucial in establishing whether transplanted cells directly improve visual function in specific retinal areas. Should such a direct impact be confirmed, subretinal transplants of stem cell-derived RPE transplants could emerge as the first effective treatment for a significant unmet medical need, potentially benefiting a large patient population.
留言 (0)