
記住我
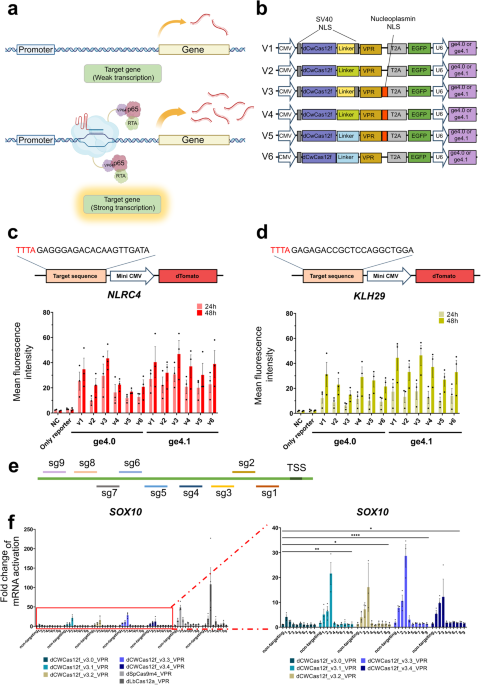


To construct an effective CRISPRa to induce the expression of target genes inside cells, we connected the trans-activator domain VPR to the dead version of the CWCas12f (dCWCas12f, D354A) system. The CRISPR-Cas12f system, in contrast to previously reported Cas12 effectors, has a relatively small size, which is pertinent for the core function of target DNA binding and cleavage within a single component [19]. Ideally, it recognizes the target DNA in a homodimeric form, allowing the recruitment of two copies of the VPR module to the upstream region of the target gene per Cas12f molecule (Fig. 1a). To optimize the operation of the dCWCas12f-VPR system within target DNA sequences, various configurations of linkers and nuclear localization signals (NLS) were employed to connect dCWCas12f and VPR (Fig. 1b; Supplementary Fig. S2). Additionally, plasmids inducing fluorescent signals within human-derived cells were co-delivered to compare their activities (Fig. 1c, d). When analyzing the activity of the dCWCas12f-VPR system in human-derived HEK293FT cells following transfection with plasmids harboring specific target sites (upstream sequence for TSS of NLRC4 and KLH29 genes) (Supplementary Table S2), normalizing the activity to GFP fluorescence intensity and comparing it to dTomato fluorescence intensity, it was observed that V3 exhibited relatively higher values (Mean fluorescence intensity 1.73 (24 h)–2.59 (48 h) for NLCR4, 0.62 (24 h)–1.43 (48 h) for KLH29), as shown in Supplementary Fig. S1. When different guide RNA versions (ge4.0 or ge4.1, Supplementary Table S1) were used [15], the ge4.1 version of the guide RNA yielded a relatively higher average fluorescence intensity (Fig. 1c, d, Supplementary Table S3). Therefore, we conducted additional follow-up experiments using the ge4.1 version of the guide RNA and the V3 version of the dCWCas12f-VPR system, which exhibited the highest value of average dTomato fluorescence for the various genes analyzed.
Fig. 1: Optimization of the dCWCas12f-based CRISPR activator for targeted transcriptional activation.
a Schematic representation of transcriptional activation system using the dCWCas12f-VPR module. b Diverse dCWCas12f-based transcriptional activator versions with different linker lengths (16, 32, and 33 amino acids) and nuclear localization signal (NLS) sequences, along with the utilization of two distinct guide RNA structures (ge4.0 and ge4.1). Mean fluorescence intensity profile generated by different versions of dCWCas12f-VPR on mini-CMV-based dTomato expression reporter system targeting NLRC4 (c) and KLH29 (d). e Schematic illustration depicting the distribution of sgRNAs for tiling assay on SOX10, relative to the transcriptional start site (TSS). f Comparative analysis of activation of endogenous gene expression in HEK293FT cells using different versions of CRISPR activator: dCWCas12f-VPR (v3.0, v3.1, v3.2, v3.3, v3.4), dSpCas9m4-VPR, and dLbCas12a-VPR. Each histogram was plotted by applying the standard error of the mean values to repeated experimental values (n ≥ 3). P-values are calculated using a two-way analysis of variance (ANOVA) and Tukey’s multiple comparisons test (ns: not significant, *P = 0.0332, **P = 0.0021, ***P = 0.0002, ****P < 0.0001). The figures were created using BioRender software (BioRender.com).
Further engineering of CRISPR-Cas12f system for enhanced regulation of gene expressionIn the previous experiment, the optimized combination of linker types and NLS with the V3 version of the dCWCas12f-VPR system, along with the ge4.1 guide RNA, was used to improve the efficiency of gene expression activation on actual genomic sites in human-derived cells (Fig. 1e, f). The introduction of amino acid mutations in a similar Cas12f system led to an increase in the targeting efficiency of Cas12f toward the target DNA sequences [18]; therefore, identical mutations ((D171R, T175R, E179A) or (D171R, T175R, K358R, E556R)) were introduced at conserved amino acid positions within the dCWCas12f-VPR system to further enhance its efficiency (Supplementary Fig. S2a). Moreover, these mutant versions were further combined with the FUS-IDR domain [28], which is known to recruit transcription initiation factors through phase separation, yielding four different versions (3.1–3.4) of the enhanced dCWCas12f-VPR system (Supplementary Fig. S2b). Using these four different versions, which were created via the introduction of various forms of mutations and FUS-IDR domain conjugation, gene expression activity in the upstream region of SOX10 in a human-derived cell line was compared and analyzed through tiling assays (Fig. 1e; Supplementary Fig. S3, Supplementary Table S2). When the 3.3 version of the dCWCas12f-VPR system was introduced into cells to induce gene expression, it consistently exhibited the highest average induction efficiency at various positions in the TSS upstream region (Fig. 1f). Additionally, this version exhibited the highest expression efficiency in the target site (sg3) of the TSS upstream region among the different versions. Notably, the enhanced 3.3 version of the dCWCas12f-VPR system (referred to as eCWCas12f-VPR) demonstrated an efficiency comparable to that of previously reported gene expression activation (CRISPRa) systems based on Cas9 or Cas12a (Fig. 1f).
Targeted activation of endogenous gene expression across multiple genes using Cas12f-based activatorWe investigated whether the previously improved eCWCas12f-VPR system could universally induce gene expression at various genomic sites (CD2, CXCR4, HBB, IL1RN, ASCL1, and HBG) in human-derived cells (Fig. 2, Supplementary Table S2). To directly compare the regulatory effects of the CRISPR-Cas module on gene expression with existing CRISPR activators, we compared it with the well-characterized Cas12a system [12]. The previously reported LbCas12a-VPR system, which is based on dLbCas12a effector derived from Lachnospiraceae bacterium recognizes a thymine-rich PAM (TTTV) similar to the PAM (TTTR) of the eCWCas12f-VPR system, enables the direct comparison of gene expression efficiency at the same genomic sites (Supplementary Fig. S4, Supplementary Table S2). Across all tested gene loci, the eCWCas12f-VPR system exhibited an overall superior gene expression amplification efficiency compared to the LbCas12a-VPR system. Application of the LbCas12a-VPR and eCWCas12f-VPR systems resulted in an average of 29.72-fold and 965.46-fold induction of gene expression, respectively. When applying the eCWCas12f-VPR system, the gene expression amplification for CD2, CXCR4, HBB, IL1RN, ASCL1, and HBG increased by an average of ~29.61-, 1.46-, 5.08-, 1.99-, 3.95-, and 242.90-fold compared to that of the LbCas12a-VPR system (Fig. 2a–f, Supplementary Table S4). Notably, for the target genes HBB, ASCL1, and HBG (Fig. 2c, e, f), for which the existing LbCas12a-VPR system was unable to induce significant gene amplification, the eCWCas12f-VPR system was effective in inducing gene expression. Similar to previous reports, for both the LbCas12a-VPR and eCWCas12f-VPR systems, we observed significant variability in the gene expression efficiency for different target DNA sequences within the TSS upstream region of each gene. Consequently, when comparing gene expression induction for the same target DNA sequences within the same gene (Supplementary Fig. S4), the eCWCas12f-VPR system demonstrated robust activity comparable to the LbCas12a-VPR system. Both systems exhibited significant efficiency variations depending on the target DNA sequence. To date, it is known that dSpCas9m4-VPR system is a standard activator for guanine-rich target sites [29]. We further compared the transcription activation efficiency of eCWCas12f-VPR to previously established dSpCas9m4-VPR activator for various gene locus (Supplementary Fig. S4). Targeted gene activation for endogenous HBG, CD2, and CXCR4 genes was conducted using dLbCas12a-VPR, eCWCas12f-VPR, and dSpCas9m4-VPR. For a rigorous comparison of activation efficiencies, specific genomic target sites were selected based on each shared PAM and protospacer characteristics (Supplementary Fig. S4a, b, e). Our results demonstrated that gene expression activation by eCWCas12f-VPR was more efficient (average of 478.37-fold increase) than well-established dSpCas9m4-VPR system (Supplementary Fig. S5). For the thymine-rich PAM (TTTV) locus, eCWCas12f-VPR showed higher or equal induction efficiency in gene expression compared to dSpCas9m4-VPR, highlighting its potential as a powerful tool for thymine-rich target gene activation.
Fig. 2: Targeted transcriptional activation with eCWCas12f-VPR across various endogenous target sites in human cell line.
Comparison of eCWCas12f-VPR and dLbCas12a-VPR for the activation of various endogenous genes in HEK293FT cells. Target genes included CD2 (a), CXCR4 (b), HBB (c), IL1RN (d), ASCL1 (e), and HBG (f). The relative value of gene activation was measured using quantitative polymerase chain reaction (qPCR) and was used to measure the fold change in mRNA expression. mRNA expression levels were normalized to the values obtained using non-targeting sgRNA (sgNT) as a control. Each histogram was plotted by applying the standard error of the mean to repeated experimental values (n ≥ 3).
Multiplexed gene activation using eCWCas12f-VPRTo enhance the potential for diverse research applications and therapeutic approaches, we further tested multiplexed gene activation with eCWCas12f-VPR system. We used a vector that co-expresses the Csy4 domain with eCWCas12f-VPR to simultaneously induce the activation of multiple target genes [30]. The vector also contains linked sgRNA expression cassettes designed with Csy4 recognition sites between each sgRNA, allowing eCWCas12f-VPR to be guided to multiple target loci respectively. After transcription, Csy4 recognizes and cleaves the Csy4 recognition site between sgRNAs. When multiple genes (CD2, CXCR4, and HBG) were targeted with eCWCas12f-VPR and multiple sgRNAs (Supplementary Fig. S6), gene expression levels for CD2, CXCR4, and HBG were upregulated by ~6.41-, 1.05-, and 7.04-fold, respectively, compared to the non-targeting control. These data indicate that using the Csy4 and eCWCas12f-VPR co-expression system, eCWCas12f-VPR can simultaneously regulate multiple target genes through the expression of multiple guide RNAs although it requires further optimization for better efficiency.
Highly specific activation of gene expression using eCWCas12f-VPRTo confirm both the effective gene expression amplification characteristics and specificity for the target genes, we compared the on/off-target gene expression profiles (on/off-target mRNA fold enrichment ratio) of the eCWCas12f-VPR system to those of the LbCas12a-VPR system (Fig. 3). We searched for common targets and respective off-target candidate sequences based on the PAM nucleotide sequences for various gene loci within human-derived cells (HBB, ASCL1, HBG, CD2) (Fig. 3a, d, g, j; Supplementary Table S5). Subsequently, we verified the induction of gene expression in both on- and off-target sequences. When the LbCas12a-VPR system was used, we observed average on/off-target ratios of 5.42-fold for HBB, 5.49-fold for ASCL1, 135.00-fold for HBG, and 298.88-fold for CD2 (Fig. 3b, e, h, k, Supplementary Fig. S7). However, when the eCWCas12f-VPR system was applied, the average on/off-target ratios were 23.18-fold for HBB, 61.60-fold for ASCL1, 11257.15-fold for HBG, and 250.71-fold for CD2 (Fig. 3c, f, i, l, Supplementary Fig. S7). When using the eCas12f-VPR system to induce gene expression, we observed an overall increase in target specificity based on the on/off-target ratios, with an average improvement of 26.06-fold when compared to the LbCas12a-VPR system. In addition to target-specific analysis, we sought to comparatively analyzed the specificity of CRISPR activators at the whole transcriptome-wide level. To this end, we performed CRISPR activation using eCWCas12f-VPR and LbCas12a-VPR constructs and conducted RNA-seq to analyze the changes of expression levels of on-target genes, HBG1 and HBG2, and corresponding off-target genes (Fig. 3m, n). We observed that both eCWCas12f-VPR and LbCas12a-VPR-based activation resulted in marked increase in HBG1 and HBG2 expression levels in RNA-seq analyses. Notably, eCWCas12f-VPR-based activation showed greater magnitude of gene activation for both HBG1 and HBG2 than any other genes (Fig. 3m). In contrary, LbCas12a-VPR-based activation resulted in activation of some potential off-target genes with higher fold difference compared to HBG1 and HBG2 (Fig. 3n). Next, we sought to quantitatively compare the specificities of eCWCas12f-VPR and LbCas12a-VPR-based activators by counting the number of genes that showed significant changes in expression levels. RNA-seq analyses showed that eCWCas12f-VPR and LbCas12a-VPR-based activation led to gene expression levels changes of 312 and 808 genes, respectively, with fold difference greater than 2 and p-value equal of less than 0.05. Together, off-target analysis in whole transcriptome-wide data suggested that eCWCas12f-VPR based activation is more specific than LbCas12a-VPR-based activation.
Fig. 3: Comparative evaluation of off-target effects induced by eCWCas12f-VPR and dLbCas12a-VPR.
Identification of expected off-target candidates for each on-target site (HBB (a), ASCL1 (d), HBG (g), and CD2 (j)) through in-silico analysis. Mismatched bases between guide RNA and the target sequence are highlighted in red for every off-target candidate sequence. Assessment of on- and off-target gene activation induced by eCWCas12f-VPR and dLbCas12a-VPR for HBB (b, c), ASCL1 (e, f), HBG (h, i), and CD2 (k, l) genes. RNA-seq analyses of eCWCas12f-VPR and dLbCas12a-VPR activators. Activated gene expression induced by eCWCas12f-VPR (m) and dLbCas12a-VPR (n) compared to the unactivated case is plotted as scatter plots, respectively. On-target genes (HBG1 and HBG2) induced by eCWCas12f-VPR and dLbCas12a-VPR were colored in red and blue, respectively.
Targeted gene activation with eCWCas12f-VPR through AAV-mediated gene deliveryTo investigate target-specific gene expression activation using eCWCas12f-VPR loaded recombinant adeno-associated virus serotype 2 (rAAV2) vectors, we designed dual rAAV2 based delivery system, one for eCWCas12f-VPR and the other for its corresponding sgRNA expression (Fig. 4a, b). Each rAAV2 was designed to express eCWCas12f-VPR under the control of the small CMVd1 promoter or sgRNA by the U6 promoter, respectively, and these purified rAAV2 particles were simultaneously transduced into HEK293FT cells. As a result, the eCWCas12f-VPR system increased HBG gene expression by 6.55-fold, demonstrating the effectiveness of the eCWCas12f-VPR system in activating endogenous genes when delivered through AAV vectors (Fig. 4c).
Fig. 4: Activation of endogenous gene expression using eCWCas12f-VPR-loaded rAAV delivery vehicles.
a eCWCas12f-VPR activator and sgRNA for eCWCas12f-VPR can be expressed under the control of CMVd1, U6 promoter, respectively. Each gene sizes required for CMVd1 promoter and eCWCas12f-VPR expression, and U6 promoter and the sgRNA gene are ~4.5 kb and 0.6 kb, respectively, which do not exceed the maximum AAV packaging limit. b Illustration of rAAV2-eCWCas12f-VPR and rAAV2-sgRNA production in HEK293FT cells and induction of target gene expression. The rAAV2 vectors containing eCWCas12f-VPR and sgRNA were purified separately to induce co-transduction and target gene activation in HEK293FT cells. c Induction of target gene (HBG) expression in HEK293FT cells by co-transduction of rAAV2-eCWCas12f-VPR and rAAV2-sgRNA particles and comparison of histograms of amplification values and NC values. NC: negative control, AAV_eCWCas12f-VPR: Co-transduction with rAAV2-eCWCas12f-VPR and rAAV2-sgRNA particles. each histogram value represents the mean value of three measurements and the error bars represent the standard error of the mean from three repeated experiments (n = 3). The figures were created using BioRender software (BioRender.com).
留言 (0)