Cloning, protein expression and purification in E. coli
Protein coding DNA sequences for METTL6 (UniProt Q8TCB7) with a 3C protease cleavable N-terminal 6×His-GST tag and SerRS (UniProt P49591) with a C-terminal 6×His-tag were cloned into a pEC vector. Point mutations of the proteins were produced by site-directed mutagenesis using single or double-mismatch primers.
All proteins were expressed in E. coli of the Rosetta II (DE3) strain and cultured in terrific broth medium. Expression was induced with 0.1 mM IPTG at 18 °C overnight. Cell pellets were frozen and kept at −80 °C before purification.
Wild type or mutant METTL6 was purified by resuspending and sonicating the cell pellet in lysis buffer containing 20 mM Tris-HCl, pH 7.5, 250 mM NaCl, 20 mM imidazole, 0.05% (v/v) 2-mercaptoethanol, 1 mM PMSF, 1 μg ml−1 DNase I, 0.5 μg ml−1 RNase and 50 μg ml−1 lysozyme. The lysate was cleared by centrifugation with 20,000g followed by filtration with a 5 μM filter and loaded on a HisTrap HP column pre-equilibrated with lysis buffer (GE Healthcare). The column was washed first with lysis buffer, then with high-salt buffer (20 mM Tris-HCl, pH 7.5, 250 mM NaCl, 1 M KCl, 50 mM imidazole) and low-salt buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 100 mM imidazole) before eluting with elution buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 300 mM imidazole). The protein was further purified by loading it on a HeparinTrap (GE Healthcare) and eluted with a gradient from 100 mM to 1 M NaCl. Protein-containing fractions were pooled, recombinant His-tagged 3C protease (EMBL PEPcore) was added and the protein was dialyzed overnight against 20 mM HEPES–KOH, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 20 mM imidazole, 0.05% (v/v) 2-mercaptoethanol. The cleaved tag and 3C protease were removed by running the dialysate over another 5 ml HisTrap column and collecting the unbound protein. The protein was concentrated using centrifugal spin concentrators (Millipore) with an MWCO of 10 kDa. As the final purification step SEC was performed on a Superdex 75 16/600 column (GE Healthcare) with 20 mM HEPES–KOH, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 1 mM dithiothreitol (DTT). Fractions with pure protein were pooled and concentrated. All protein purification steps were carried out at 4 °C.
SerRS was purified by resuspending the cell pellet in lysis buffer (20 mM Tris-HCl, pH 7.5, 250 mM NaCl, 20 mM imidazole, 0.05% (v/v) 2-mercaptoethanol, 1 mM PMSF, 1 μg ml−1 DNase I, 0.5 μg ml−1 RNase and 50 μg ml−1 lysozyme) and lysed by microfluidization with a pressure of 275 kPa. The lysate was cleared by centrifugation at 20,000g followed by filtration with a 5 μM filter before loading on a HisTrap HP column (GE Healthcare). The column was washed first with lysis buffer, then with high-salt buffer (20 mM Tris-HCl, pH 7.5, 250 mM NaCl, 1 M KCl, 50 mM imidazole) and low-salt buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 50 mM imidazole) before eluting with elution buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 300 mM imidazole). The eluted protein was then applied to a 5 ml Q-trap (GE Healthcare) and eluted with a gradient from 100 mM to 1 M NaCl. Pure protein-containing fractions were pooled and concentrated using centrifugal spin concentrators with an MWCO of 30 kDa (Millipore). Finally, SEC was performed on a Superdex 200 10/300 column with 20 mM HEPES–KOH, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 1 mM DTT. Fractions containing the pure protein were pooled and concentrated. The purified proteins were flash-frozen in liquid nitrogen and stored at −80 °C for further experiments.
Cryo-EM sample preparation
The DNA sequences coding for SerRS and METTL6 were cloned into a psLIB vector by Gibson Assembly, yielding a fusion construct with the sequence for METTL6 fused directly to the C terminus of SerRS with a C-terminal 3C protease cleavable EGFP tag. The fusion protein was expressed in Hi5 cells. After 72 h of protein expression at 25 °C, the cells were collected by centrifugation, resuspended in lysis buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 0.05% (v/v) 2-mercaptoethanol, 1 mM PMSF) and lysed by sonication. The lysate was cleared by centrifugation with 20,000g at 4 °C followed by filtration with a 5 µM filter. All subsequent purification steps were carried out at 4 °C.
The lysate was incubated with 1 ml EGFP nanobody resin for 30 minutes. The resin was washed two times with a wash buffer containing 20 mM Tris-HCl, pH 7.5, 100 mM NaCl and 2 mM MgCl2. The SerRS–METTL6 fusion construct was eluted from the resin by cleavage of the EGFP tag using the 3C protease cleavage site: 500 µl wash buffer with 0.08 mg ml−1 3C protease (EMBL PEPcore) was added to the resin and incubated for approximately 2 h. The cleaved protein was removed from the resin, filtered with a Spin-X centrifuge tube filter (Costar) with a pore size of 0.22 µM and concentrated using a centrifugal spin concentrator (Millipore) with an MWCO of 10 kDa. As the final purification step, the protein was purified by SEC on a Superdex 200 3.2/300 column equilibrated in 20 mM HEPES–KOH, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 0.5 mM TCEP. Fractions containing the fusion construct bound to tRNA were frozen with liquid nitrogen and stored at −80 °C until grid preparation.
The sample was diluted to 0.2 mg ml−1 with a buffer containing 20 mM HEPES–KOH (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 0.5 mM TCEP and 1 mM sinefungin. For electron microscopy grid preparation, UltrAuFoil grids with an R 1.2/1.3 300 mesh (EMS) were glow-discharged on both sides for 40 seconds with 30 mA at 0.45 bar using a Pelco EasyGlow glow-discharging device. Then, 2 µl of the diluted sample was applied to each side of the grid and the sample was vitrified in liquid ethane using a MARK IV Vitrobot (FEI) with the following settings: 100% humidity, 4 °C, 3 seconds blot time and a blot force of 0.
Cryo-EM data acquisition, processing and model building
Data were acquired on a Titan Krios transmission electron microscope (FEI) equipped with a 300 kV accelerating voltage field emission gun electron source, a Quantum energy filter (Gatan) and a Gatan K3 camera, at ×130,000 magnification with a pixel size of 0.645 Å per pixel. The microscope was operated using the software SerialEM67. Videos were acquired in counting mode with an electron dose of 63.27 e−/Å2 in 40 frames with a defocus range from −0.8 µm to −1.8 µm.
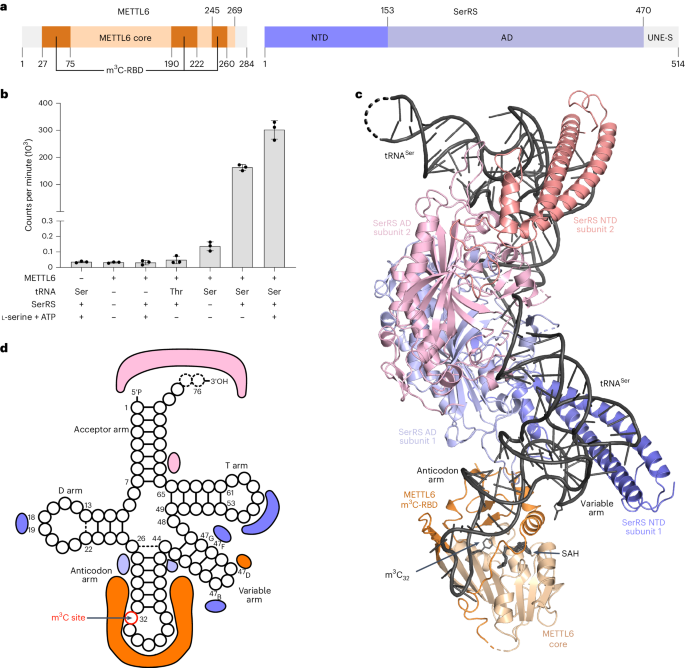
Motion correction and contrast transfer function (CTF) estimation on the collected micrographs were performed using RELION v.3.1 (ref. 68). Particles were picked with the BoxNet_20220403_191253 model in Warp69, and extracted in RELION in a 550-pixel box. A total of 1,625,946 particles were used as input for data processing using cryoSPARC70. After an initial round of two-dimensional (2D) classification to remove bad particles and particles containing only tRNA, the remaining 1,282,076 particles were used for several rounds of ab initio 3D classifications, which resulted in different classes containing dimeric SerRS with one or two tRNA molecules and 0–2 copies of METTL6 bound (Extended Data Figs. 2 and 3 and Table 2). The particles of the 3D class with dimeric SerRS, two tRNAs and one molecule of METTL6 were subjected to several rounds of CTF refinement and Bayesian polishing in RELION v.3.1. The refined particles were re-imported into cryoSPARC to perform a final nonuniform refinement71. All reported resolutions were determined using the gold standard FSC method implemented in cryoSPARC, with a cutoff of 0.143.
The highest resolution model was built in Coot using the available crystal structures of tRNA-bound human SerRS (PDB 4RQE)37 and of METTL6 (this study) as starting templates. The model was refined by several rounds of real-space refinement in Phenix (v.1.16) and manual refinement in Coot (v.0.8.9.2). The other models were built based on the highest resolution model, with minimal refinement. Illustrations were generated with ChimeraX66, maps shown at a level of 0.3, and PyMOL72, maps shown at a σ level 0.18.
Protein crystallography
METTL6 was crystallized by vapor diffusion in the hanging drop format at 20 °C at a concentration of 4 mg ml−1 supplanted with 2 mM SAH and 5 μM TCEP. A 1 μl portion of protein solution was added to 1 μl of reservoir solution containing 0.1 M Bis-Tris propane, pH 6.5, 0.2 M sodium sulfate and 20% PEG 3350. After two weeks crystals were soaked for two minutes in a cryoprotectant solution containing 2 mM SAH, 0.1 M Bis-Tris propane, pH 6.5, 0.2 M sodium sulfate, 10% PEG 400 and 30% PEG 3350 and flash-frozen in liquid nitrogen.
A truncated construct of METTL6 consisting of residues 40 to 269 was crystallized at a concentration of 5 mg ml−1 supplanted with 1 mM SAH in the sitting drop format platform by adding 100 nl protein solution to 100 nl of a reservoir solution containing 0.1 M Bis-Tris, 0.2 M ammonium sulfate and 25% PEG 3350 at 20 °C. The crystals were collected using the automated Crystal Direct collecting system of the HTX platform73 and flash-frozen without cryoprotectant.
X-ray diffraction datasets of full-length METTL6 were collected on the MASSIF-1 beamline at the ESRF with an X-ray energy of 12.65 keV (0.9801 Å). For solving the phase problem for METTL6 by single-wavelength anomalous dispersion (SAD) with the anomalous signal from sulfur, a total of 35 X-ray diffraction datasets with an X-ray energy of 6 keV (2.066 Å) were collected on beamline P13 at the PETRA-III storage ring at DESY. X-ray diffraction datasets of METTL6Δ were also obtained on this beamline with an X-ray energy of 12.3 keV (1.008 Å).
All diffraction images were processed using XDS74. To solve the phase problem, five of the 6-keV datasets were clustered and merged using BLEND75 and fed into the CRANK2 pipeline for SAD phasing75, which resulted in a low-resolution (4.42 Å) structure of METTL6. This low-resolution structure was used to obtain phases for the other datasets by molecular replacement using Phenix-PHASER76. For all models, several rounds of model building using Coot77 and data refinement using Phenix-REFINE78 were performed. Ramachandran statistics for the METTL6 (full length) are 97.42% favored, 2.58% allowed and 0% disallowed, and for the METTL6 (40–269) are 95.18% favored, 4.82% allowed and 0% disallowed. Data collection and refinement statistics are shown in Table 1.
tRNA in vitro transcription
The DNA sequences coding for a T7 promoter, human tRNASerUGA or tRNAThrCGU, respectively, directly followed by a BstN1 cleavage site, were cloned into a pUC19 vector. The vector was linearized using BstN1 and used as a template for runoff transcription with T7 polymerase. A 1 mg portion of linearized DNA was transcribed in vitro in a reaction containing 40 mM Tris-HCl, pH 8.0, 30 mM MgCl2, 5 mM DTT, 1 mM spermidine, 0.01% (w/w) Triton X100, 4 mM of each NTP and 50 µg ml−1 recombinant T7 polymerase (EMBL PEPcore), which was incubated for 16 h at 37 °C. The produced RNA was precipitated with isopropanol, purified via urea–PAGE, desalted using a Pierce dextran desalting column (ThermoFisher) and concentrated using a centrifugal spin concentrator (Millipore) with a MWCO of 10 kDa. The tRNA was refolded by heating it for 2 minutes to 95 °C before adding MgCl2 to a final concentration of 2 mM and quickly placing it on ice.
Thermal shift assay
METTL6 was diluted to a final concentration of 20 μM in a buffer containing 20 mM HEPES–KOH (pH 7.5), 100 mM NaCl, 2 mM MgCl2, 1 mM DTT, 5× SYPRO Orange (Invitrogen) and 1 mM of cofactor (SAM, SAH or sinefungin). Fluorescence was measured in a real-time PCR machine (Stratagene Mx3005P) while subjected to a temperature gradient from 24.6 °C to 95°°C in steps of 1 °C and 1 min.
Fluorescence polarization assay
The in-vitro-transcribed tRNASerUGA and tRNAThrCGU were labeled on the 3′ end with fluorescein. The DNA and RNA oligomers (labeled on the 3′ end with 6-carboxyfluorescein) were purchased from biomers.net. All nucleic acids were refolded as described above. The labeled tRNAs with a concentration of 25 nM were incubated with METTL6 of different concentrations in a total volume of 20 µl for 15 minutes at room temperature in a 384-well flat black microplate (Greiner) in a buffer of 20 mM HEPES–KOH (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 0.1% Tween, 1 mM DTT, 0.5 U µl−1 RNasin and 0.5 mM sinefungin. Fluorescence polarization was measured at a temperature of 25 °C with a ClarioStar plate reader (BMG Labtech) with an excitation wavelength of 460 nm and an emission wavelength of 515 nm. Data analysis was performed using GraphPad Prism using the single site binding fit.
Size-exclusion complex formation assay
METTL6, SerRS and tRNASerUGA (each at a concentration of 30 µM; METTL6 supplemented with 1 mM sinefungin to stabilize the complex) were incubated for 10 minutes on ice before subjecting them to analytical SEC using a Superdex 200 Increase 3.2/300 column (GE Healthcare) equilibrated in a buffer composed of 20 mM HEPES–KOH, pH 7.5, 50 mM NaCl, 2 mM MgCl2, 0.5 mM TCEP. Fractions were analyzed via SDS–PAGE stained with Coomassie blue and urea–PAGE stained with methylene blue for protein and RNA content, respectively.
Methyltransferase assay
Methyltransferase assays were performed in 6 mM HEPES–KOH (pH 7.9), 0.4 mM EDTA, 10 mM DTT, 80 mM KCl, 1.5 mM MgCl2, RNasin (40 U µl−1) (Promega) and 1.6% glycerol in a total volume of 200 µl. Proteins (METTL6, SerRS) were used in 0.05 µM final concentration. As substrate, 0.4 µM of in-vitro-transcribed tRNA(Ser) or tRNA(Thr), or 5 µg of total RNA extracted from HeLa cells, was added. In addition, 4 µM of serine (Sigma) and 2 mM of ATP were added where indicated. A 1 µl portion of 3H-labeled SAM (1 mCi ml−1; PerkinElmer) was added to the mixture and then incubated at 30 °C for 1 h with gentle shaking. Then, another 1 µl of 3H-SAM was added and the assay continued at room temperature (22 °C) overnight followed by column purification of the RNA with the Zymo Research RNA Miniprep kit (used according to the manufacturer’s instructions). After elution of RNA from the columns in 150 µl nuclease-free H2O, 500 µl of AquaLight liquid scintillation counter cocktail (Hidex) was added and tritium incorporation analyzed by scintillation counting with a Triathler Counter (Hidex). All data of in vitro methyltransferase assays are shown as blank-subtracted mean of three scintillation counts and three experimental replicates that were carried out for each experiment.
Sample preparation for LC–MS/MS and detection of modified nucleosides
The Trichoplusia ni tRNA that copurified with the SerRS–METTL6 fusion construct was isolated via chloroform–phenol extraction and precipitated with ethanol and sodium acetate. Around 300 ng of the tRNA was digested in an aqueous digestion mix (30 μl) to single nucleosides by using 2 U alkaline phosphatase, 0.2 U phosphodiesterase I (VWR) and 2 U benzonase in Tris (pH 8, 5 mM) and MgCl2 (1 mM) containing buffer. Furthermore, 0.5 μg tetrahydrouridine (Merck), 1 μM butylated hydroxytoluene and 0.1 μg pentostatin were added. After incubation for 2 h at 37 °C, 20 μl of LC–MS buffer A (QQQ) was added to the mixture. A stable isotope labeled SILIS (gen² (ref. 79)), was added to each replicate and calibration solution of synthetic standards before injection into the QQQ MS.
LC–MS/MS of nucleosides
Modified nucleosides were identified and quantified using mass spectrometry. An Agilent 1290 Infinity II equipped with a diode-array detector combined with an Agilent Technologies G6470A Triple Quad system and electrospray ionization (ESI) mass spectrometer (Agilent Jetstream) was used.
Nucleosides were separated using a Synergi Fusion-RP column (Synergi 2.5 μm Fusion-RP 100 Å, 150 mm × 2.0 mm, Phenomenex). LC buffer consisting of 5 mM NH4OAc, pH 5.3 (buffer A) and pure acetonitrile (buffer B) was used. The gradient started with 100% buffer A for 1 min, followed by an increase to 10% buffer B over a period of 4 min. Buffer B was then increased to 40% over 2 min and maintained for 1 min before switching back to 100% buffer A over a period of 0.5 min and re-equilibrating the column for 2.5 min. The total time was 11 min and the flow rate was 0.35 ml min−1 at a column temperature of 35 °C.
An ESI source was used for ionization of the nucleosides (Agilent Jetstream). The gas temperature (N2) was 230 °C with a flow rate of 6 l min−1. Sheath gas temperature was 400 °C with a flow rate of 12 l min−1. Capillary voltage was 2,500 V, skimmer voltage was 15 V, nozzle voltage was 0 V and nebulizer pressure was 40 psi. The cell accelerator voltage was 5 V. For nucleoside modification screening, MS2Scan (m/z 250–500) was used. For quantification, a DMRM and positive-ion mode was used (Supplementary Table 2).
Data analysis of nucleosides
For calibration, synthetic nucleosides were weighed and dissolved in water to a stock concentration of 1–10 mM. Calibration solutions ranged from 0.0125 pmol to 100 pmol for each canonical nucleoside, and from 0.00625 pmol to 5 pmol for each modified nucleoside. Analogous to the samples, 1 µl SILIS (10×) was co-injected with each calibration. The calibration curve and the corresponding evaluation of the samples were performed using Agilent’s quantitative MassHunter software. All modification abundances were normalized to the amount of RNA injected using the sum of all canonicals.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.


留言 (0)