
記住我
Cardiovascular disease (CVD) due to atherosclerosis is the foremost cause of premature mortality worldwide. It is generally accepted that elevated blood lipids play an important role in the development of atherosclerotic plaques and cardiovascular disease etiopathogenesis. Over the last two decades, the field of lipid study has paid attention to the study of proprotein convertase subtilisin/kexin type 9 (PCSK9). PCSK9 is one of the three most important genes involved in familial hypercholesterolemia, other than LDLR or apolipoprotein B (APOB). While PCSK9 gain-of-function (GOF) mutations associated with elevated blood lipids, carriers of loss of-function (LOF) mutations benefited from low-density lipoprotein cholesterol (LDL-C) reduction by up to 28%, accompanied by an 88% reduced risk of coronary artery disease [1]. Hepato-specific reduction LDL-C levels remains the first strategy in managing patients with familial hyperlipidemia and those with clinical atherosclerotic cardiovascular disease not reaching lipid-reducing goals [2]. To mimic PCSK9 natural inhibition, in 2015 PCSK9 monoclonal antibodies, the first strategy to inhibit PCSK9 therapeutically successfully approved in addition to statin therapy. More recently, silencing RNA (siRNA) reduced LDL-C by 45–60% [3]. Here, we systematically reviewed the available scientific evidence of genetic variation on PCSK9 and assessed the efficacy of PCSK9 inhibitors in cardiovascular outcomes, including cardiovascular death, myocardial infarction, and stroke.
PCSK9: Biological FunctionPCSK9, the ninth member of the proprotein convertase family, is a serine protease that caught the attention of the scientific community in 2003 when the discovery of the first natural mutants of PCSK9 revealed the implication of an as-yet-unknown actor in cholesterol homeostasis [4, 5]. PCSK9 is mainly expressed on hepatocytes surface and has been shown to act both intracellularly playing a role as a chaperone in the degradation of the LDL receptor (LDLR), as well as a secreted factor promoting LDLR internalization from the hepatocellular surface [6]. PCSK9 regulates the degradation of the LDLR in response to cholesterol levels within the cell [7].
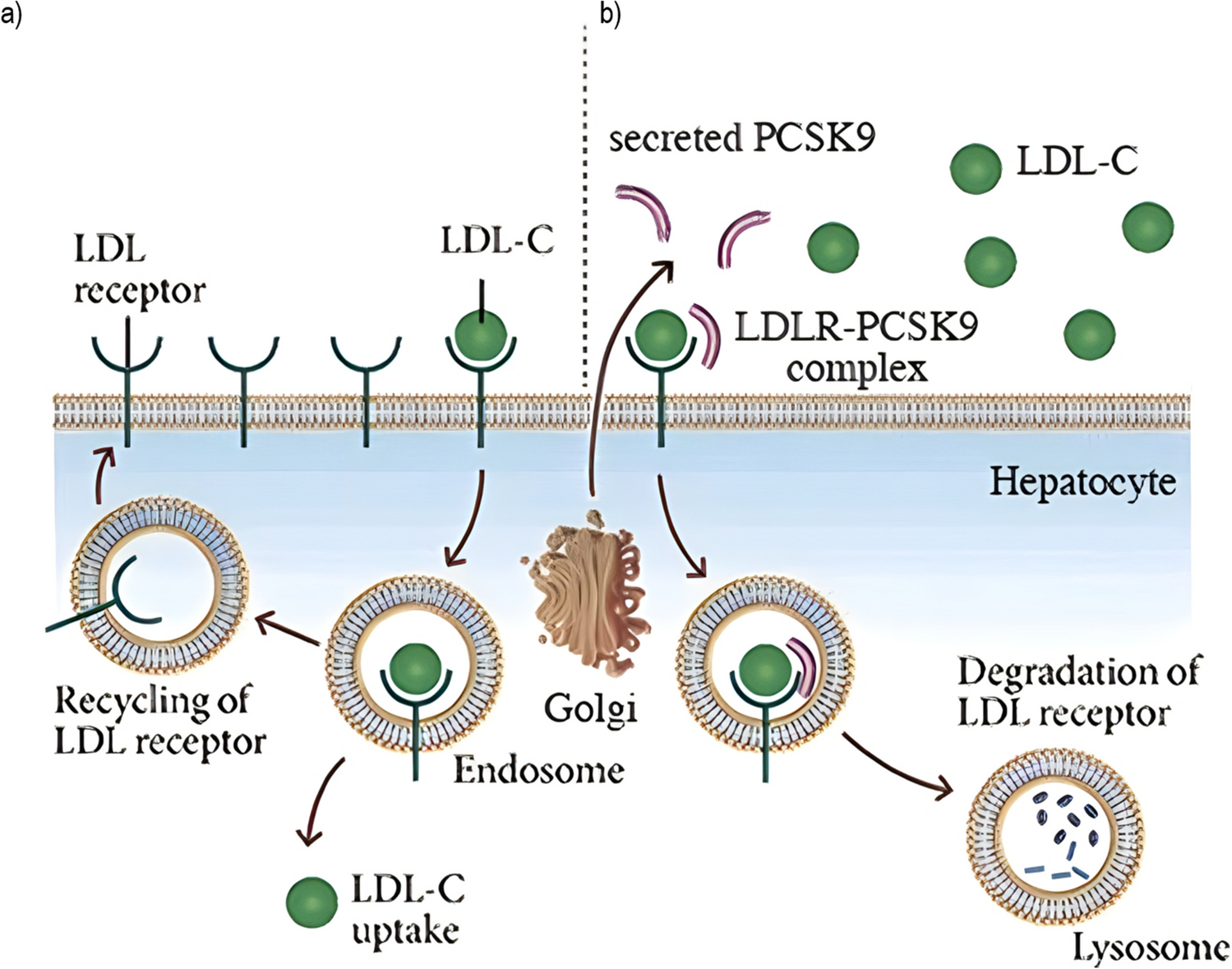
PCSK9 protein structure is characterized by a signal sequence, a prodomain, and a catalytic domain, followed by a C-terminal region. PCSK9 is synthesized as an inactive 75 kDa proenzyme that undergoes autocatalytic cleavage in the endoplasmic reticulum (ER) which produces an approximately 60-kDa catalytic fragment. Autocatalytic cleavage of the zymogen in the ER is essential for its transport from this compartment and for its secretion. PCSK9 favors LDLR degradation independently of its catalytic activity by involving mainly extracellular and possibly intracellular pathways [8]. PCSK9 catalytic domain (aa153–421) strongly interacts with the EGF-A domain (aa314–355) of the LDLR (extracellular domain, ECD). This prevents the LDLR from forming a closed conformation, making the receptor susceptible to enzymatic degradation, rather than being recycled to the cell surface (Fig. 1) [9].
Fig. 1
a LDL-C uptake and recycling of LDLR. b PCSK9-mediated degradation of LDLR. The protein PCSK9 regulates the number of LDL receptors (LDL-Rs) on a cell’s surface. When PCSK9 binds to these receptors, the receptors do not get recycled but are broken down in cellular compartments called lysosomes instead. Adapted with permission from Publisher [10]
PCSK9 Genetic VariantsThe human PCSK9 gene is located on chromosome 1p32, and consist of 12 exons, which encode 692 amino acid protein (NP_777596.2). PCSK9 gene is highly polymorphic with the distribution of genetic variants in all its domains affecting PCSK9 synthesis, secretion, and activity [11,12,13] (Fig. 2). In fact, gain-of-function (GOF) mutations occurred less frequent than loss-of-function (LOF) mutations.
Fig. 2
Number of genetic variants in PCSK9 (transcript ID ENST00000302118.5) based on their functional effect: a distribution of mutations per domain; b types of mutation [11]
Major findings confirmed the significant impact of PCSK9 genetic variants on plasma LDL-C concentrations by affecting the LDLR pathway [4, 14, 15]. Beyond LDLR, PCSK9 affects also other lipoproteins, lipoprotein(a) [Lp(a)], and HDL-cholesterol levels are associated with circulating PCSK9 levels [16,17,18,19]. PCSK9 may also play a role in the postprandial phase by modulating triglyceride-rich lipoprotein (TGRL) metabolism [20] (Supplementary file 1).
Gain-of-Function MutationsCarriers of GOF PCSK9 mutations over-express PCSK9, and therefore have very high plasma levels of LDL-C due to increased LDLR degradation. Several GOF missense mutations have been associated with hypercholesterolemia and premature atherosclerosis with overt CVD, such as myocardial infarction (MI) and stroke (S127R; F216L, D374Y, N157K; E32K; E228K; F216L, R357H, R215H, R496W, N35Y) [4, 5, 14, 15, 21,22,23,24,25,26,27].
Variant p.Ser127Arg interferes with autocatalytic cleavage, which is crucial for secretion from the cell. The p.Arg218Ser, p.Phe216Leu, and p.Asp374Tyr mutations result in total (p.Arg218Ser) or partial loss of the furin/PC5/6A processing of PCSK9, which increases the stability of PCSK9 [28, 29]. PCSK9 S127R GOF mutation carriers demonstrated affected TG and LDL metabolism with a threefold increased production rate of apoB-100, compared with controls or LDLR-mutated patients, which led to direct overproduction of VLDL (threefold), intermediate-density lipoprotein (IDL, threefold), and LDL (fivefold) [30].
Elevated fasting plasma TG levels and increased cardiovascular risk were confirmed in carriers of another GOF PCSK9 mutation E670G (− 23968A > G, rs505151) located in exon 12 [12, 31]. PCSK9 E670G polymorphism is mainly associated with some serum lipid parameters in the Han population. The G allele carriers had higher serum HDL-C and apoAI levels in males, lower serum apoB levels, and higher apoAI/apoB ratio in females than the non-carriers of the G allele [32]. Interestingly, when PCSK9 mutations were investigated in a Canadian Caucasian population, carriers of the E670G variant showed a non-significant difference in serum PCSK9 but a significantly lower TG concentration (27.7%; p = 0.039), not observed even for LOF mutations [33]. In a Japanese population, PCSK9 GOF E32K was associated with over 30% increased plasma levels of PCSK9, whereas these results were confirmed also by media from transiently transfected HepG2 cells, compared to controls. In patients, homozygotes for the E32K mutation was a twofold higher LDL-C levels always present with a mutation in LDLR, and in the heterozygous carriers, almost half the effect was observed. This could suggest a PCSK9 E32K effect on LDL-C levels via increased mass and function of PCSK9 and could exacerbate the clinical phenotypes of patients carrying LDLR mutations [23]. Other PCSK9 GOF mutants (F216L, R357H, and R215H) were described in different FH French families with tendon xanthomas, CHD, premature MI, and stroke [4, 5, 22]. GOF mutation in catalytic domain R215 was described to segregate with hypercholesterolemia in the Norwich family [24]. D374H was detected in one Portuguese proband and R496W in an Italian proband, together with E228K in LDLR [25, 26]. Another study demonstrated that variant p.Leu108Arg exhibited a ∼twofold-enhanced degrading activity towards the LDLR, resulting in a GOF, while variant p.Asp35Tyr created a novel Tyr-sulfation site, which may enhance the intracellular activity of PCSK9 [14]. GOF mechanism could represent a higher activity of the LDLR-degrading function of PCSK9. However, a more recent hypothesis suggests that post-translational modifications within residues 31–60 may affect the inhibitory activity of this segment and represent a mechanism for fine-tuning the activity of PCSK9 towards the LDLR. GOF mutations in the catalytic domain, which involve charged residues, could affect the positioning of this segment [27].
Another GOF variant G516V (c.1547G > T) was found in five index patients, and cascade screening identified 15 additional carriers. LDL-C levels were higher in those 15 carriers compared with the 27 non-carriers (236 ± 73 versus 124 ± 35 mg/dL; P < 0.001). In vitro studies demonstrated the pathogenicity of the G516V variant. Analysing LDL-C differences using generalized estimating equations revealed that the differences between carriers and non-carriers exceeded 39 mg/dL only for the G516V mutation [34].
Loss-of-Function MutationsContrariwise, genetic PCSK9 deficiency has been strongly associated with low plasma cholesterol levels and decreased cardiovascular risk, clearly demonstrated from several observational, randomization, and clinical studies [1, 12, 35,36,37,38,39,40]. Most of the LOF mutations result from either a deficiency in the synthesis or in the secretion of the PCSK9 protein, due to failure to exit the endoplasmic reticulum or failure to undergo the autocatalytic cleavage. Some LOF mutations could dramatically lower plasma PCSK9 up to 79%, and result in no immuno-detectable circulating PCSK9 levels (R104C/V114A, C679*; Y142X/290_292delGCC; Y142X/C679X; Q619X, Q679X, Y142X). However, these null carriers appeared healthy and fertile, with no observable signs of illness due to the absence of PCSK9. PCSK9 null compound heterozygotes did not differ from heterozygotes in any other cardio-metabolic trait, notwithstanding lower median LDL-C [13, 41,42,43,44].
Mendelian randomization studies have showed that PCSK9 genetic variants linked with low LDL-C levels were associated with reduced cardiovascular and all-cause mortality. For instance, the Copenhagen Population Study and Copenhagen City Heart Study have shown a 1 mmol/L (38.7 mg/dL) reduction in LDL-C levels due to the carriage of PCSK9 variants (R46L, R237W, I474V, and E670G) was associated with a 67% reduction in cardiovascular death (risk ratio 0.33, 95% confidence interval 0.19–0.58; p < 0.001) and a 28% reduction in all-cause death (risk ratio 0.72, 95% confidence interval 0.60–0.88; p = 0.001) [45].
LOF R46L represents one of the low-frequent PCSK9 variants (the MAF of the T allele ranged from 1 to 3.2%; 4.8% in a French–Canadian population). Despite its more moderate LDL-lowering effect (9 − 15%), the PCSK9 R46L allele was associated with a significant reduction in the incidence of CHD (47%) [1]. Homozygous R46L carriers showed a decreased trend of fasting TG levels (115 mg/dL) and higher HDL levels (70 mg/dl). In FH subjects carrying the 46L allele, significantly decreased apo-B and non-HDL concentrations with less severe clinical features were observed [46]. Interestingly, PCSK9 R46L carriers were observed an allele-dependent lower effect on lipoprotein(a) (Lp(a)) levels versus non-carriers (9 mg/dL in heterozygote, 8 mg/dL in homozygous vs. 10 mg/dL in non-carriers). The application of results regarding the link among PCSK9, LDLR, and Lp(a) metabolism is questionable, especially since Lp(a) plasma levels differ among different ethnic groups [47,48,49].
There is also evidence that PCSK9 LOF R46L according to apoE genotype would reveal some metabolic relationships. TG concentrations decreased significantly in the apoE3/E3 carriers with the R46L mutation with invariable plasma free-fatty acid (FFA) concentrations, compared with the apoE3/E2 and apoE3/E4 carriers. Subjects with both the R46L and apoE3/E2 genotypes showed a tendency toward insulin resistance, as indicated by a twofold increase in insulin, and leptin concentrations, compared with those without apoE3/E2 [39]. Emerging evidence suggests that PCSK9 LOF may also influence glucose homeostasis and insulin sensitivity. Beyond cardiovascular benefits, LOF R46L may increase the risk of diabetes in individuals with impaired fasting glucose levels [50]. Furthermore, carriers of PCSK9 and insertion of insLEU within positions 15 to 21 of the signal peptide of PCSK9 showed also increased occurrence of prediabetes and diabetes status [51]. Another study on animal models showed that glucose clearance is significantly impaired in PCSK9 KO mice fed a standard or a high-fat diet for 20 weeks compared with wild-type animals without affecting insulin sensitivity. PCSK9 KO mice presented larger islets with increased accumulation of cholesteryl esters, paralleled by increased intracellular insulin levels and decreased plasma insulin and C-peptide levels. This phenotype was completely reverted in PCSK9/LDLR DKO mice, implying the LDLR as PCSK9 target responsible for the phenotype observed [52]. Interestingly, results from the genetic and preclinical studies showing an increased risk of diabetes do not extrapolate to randomized trials results, but definitive long-term data are still lacking.
PCSK9 R46L variant is also associated with a twofold increased prevalence of hepatic steatosis and higher epicardial adiposity in human carriers of the PCSK9 R46L mutation. A similar observation was recapitulated in PCSK9 KO mice, showing increased visceral adipose tissue compared to the native genetic form [18, 20].
Postprandial studies have shown that in vivo PCSK9 deficiency was associated with a twofold decrease in postprandial TG levels [16]. In two heterozygous carriers of LOF PCSK9 mutation R104C-V114A, no alteration in TG compared with non-carriers in the postprandial state, and also no change in PCSK9 levels following an oral fat load in a small ten-patient sample was present [53]. Another study showed that carriers of LOF variants A53V, I474V, and/or R46L had no differences in PCSK9 levels, significantly lower LDL-C levels, and slightly lower TG levels in the fasting state versus non-carriers. Postprandial, PCSK9 and LDL-C levels are decreased in LOF carriers vs. non-carriers, but TG levels raised significantly to similar levels after the oral fat load. Interestingly, the same author observed that LOF PCSK9 leads to an inhibitory action on adipocyte differentiation in vitro, in both fasting and postprandial states. Furthermore, PBMC from PCSK9 LOF variant subjects showed significantly increase mRNA levels of some pro-inflammatory markers after meal [24]. Moderate postprandial lipemia was observed in LOF carriers compared to non-carrier controls after an oral fat load. It is of note that LOF was considered for carriers of the L10ins/A53V and/or I474V and/or R46L mutations, without significant effect on PCSK9 plasma levels. Moreover, as TG, apoB48, and VLDL-C were already lower compared to non-carrier controls (a phenotypic trait not observed in LOF hit variants), it remains to be clarified whether the observed effect should be consistently attributed to PCSK9 deficiency per se. In any case, there was no significant difference in fasting HDL-C (p = 0.46), apoCII (p = 0.13), and apoCIII (p = 0.66) between the groups, or the CII/CIII ratio between groups, suggesting that alterations in lipolysis are less likely a mechanism for PCSK9’s action on TGRL clearance in studied population [54, 55].
PCSK9 Inhibitors and Cardiovascular OutcomesVarious approaches have been tried to mimic natural PCSK9 inhibition, such as FDA-approved monoclonal antibodies (mAb) and non-antibody approaches including small interfering RNA (siRNA). Other approaches use therapeutic genome editing via CRISPR/Cas9 technologies (clustered regularly interspaced short palindromic repeats), a base editor which promises prolonged or even permanent reduction in circulating LDL-C levels, small molecule inhibitors (peptides/adnectins), gene silencing using antisense oligonucleotides (ASO), or peptide-based anti-PCSK9 vaccination [56,57,58]. Recently, the first oral PCSK9 inhibitor, macrocyclic peptide MK-0616, was designed to lower LDL-C via the same biological mechanism as currently approved injectable PCSK9, and it has already demonstrated statistically significant reductions in LDL-C up to 60.9% in phase 2b randomized trial [59]. Moreover, the current pre-clinical evidence regarding novel mechanisms for LDL-C lowering reveals annexin A2 as a natural extrahepatic inhibitor of the PCSK9, while depletion of protein denitrosylase SCoR2 (S-nitroso-coenzyme A reductase 2; AKR1A1) in mice lowers serum cholesterol by inhibiting liver secretion of PCSK9 [60, 61].
Among the therapeutic interventions targeting PCSK9 that are currently in use, PCSK9-mAb evolocumab (Repatha®, Amgen) and alirocumab (Praluent®, Sanofi-Ave
留言 (0)