Reagents
Monoclonal antibodies specific for NLRP1 (NBP1-54899), NLRP3 (NBP2-12446) and p21/CIP1/CDKN1A (NBP2-29463) were purchased from Novus Biologicals (Colorado, USA). Similarly, anti-Caspase 1, IL-1β (D3U3E), cGAS (#79978), p16INK4A (mouse: ab189034 and human: 18769S) were obtained from Cell Signaling Technology (Beverly, MA, USA). Finally, IL-6 (sc-32296), p53 (sc-126) antibodies and DAPI were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat Anti-Rabbit IgG H&L (HRP), goat Anti-Mouse IgG, H&L Chain Specific Peroxidase Conjugate, Poly(deoxyadenylic-thymidylic) acid sodium salt (Poly dA-dT), BSA and Triton X-100 were obtained from Merck (Darmstadt, Germany). Val-boroPro—Calbiochem 5314650001 was obtained from Merck (Darmstadt, Germany). Necrosulfonamide was obtained from Sigma-Aldrich (Saint Louis, USA). A cocktail of protease inhibitors (Complete™ Protease Inhibitor Cocktail) was purchased from Boehringer Mannheim (Indianapolis, IN). The Immun Star HRP substrate kit was obtained from Bio-Rad Laboratories Inc. (Hercules, CA). Secondary Alexa Fluor 488 Goat Anti-Rabbit Antibody was obtained from Thermo Fisher (MA, USA). Finally, siRNAs of control and cGAS (AM16708 129125), NLRP3 (4392420 S41555) and NLRP1 (4392420 S22520) were obtained from Invitrogen (Eugene, OR, USA).
Ethical statements
Animal studies were performed in accordance with the European Union guidelines (2010/63/EU) and the corresponding Spanish regulations for the use of laboratory animals in chronic experiments (RD 53/2013 on the care of experimental animals). All experiments were approved by the local institutional animal care committee.
Animals
For all experiments, only male mice were used. WT C57/BL6/J, Nlrp1a−/−Nlrp1b−/−Nlrp1c−/− (Nlrp1−/−) (C57BL/6J background, provided and originally generated and characterized in the laboratory of Seth L Masters, reference [10]) and Nlrp3−/− transgenic mice (C57BL/6J background, provided by Bernhard Ryffel and originally generated and characterized in the laboratory of J. Tschopp, reference [11]), weighing 25–30 g, were maintained on a regular 12 h light/dark cycle. All groups had ad libitum access to their prescribed diet and water throughout the study. Body weight and food intake were monitored weekly. Animal rooms were maintained at 20–22 °C with 30–70% relative humidity.
Irradiation
At 5–6 months of age, mice were sub-lethally irradiated (NDT 320 or X-RAD225, 225 kV) with a total of 12 Gy of X-ray irradiation (3 times 4 Gy, with 2 days recovery between doses). Two days prior to irradiation (IR), and for 14 days post-IR, mice received 1% Baytril solution (Broad-spectrum antibiotic) in drinking water. At 1 month after IR, mice were sacrificed at the end of the study by cervical dislocation and tissues harvested, and stored in 4% paraformaldehyde for 24 h for paraffin embedding, or frozen in liquid nitrogen. Blood samples were isolated by cardiac puncture.
Histological study
After sacrifice of mice, livers were excised and immediately stored in 4% paraformaldehyde at room temperature for 24 h for paraffin embedding after a brief rinse with PBS. The specimens were cut into 5-μm sections and stained with hematoxylin and eosin. The images were evaluated by a pathologist to find possible damages.
Immunofluorescent staining of paraffin-embedded sections
Paraffin sections were attached to superfrost plus slides (Menzel-Glaser, Braunschweig, Germany) at 60 °C for 1 h. Deparaffinization was performed by pure xylol washes 3 times for 10′ each. Slides were rehydrated by ethanol solutions immersion (from 100 to 70%) for 5′ each and rinsed with deionized water. For the heat antigen retrieval, slides were immersed in sodium citrate 10 mM (unmasking solution) and microwaved at 800 W for 15′, then samples were kept at room temperature until cool down. Slides were rinsed with PBS 1 × 3 times, and then blocking solution (2% BSA, 0.05% Triton X-100 in PBS 1×) was applied for 1 h. The samples were surrounded with a hydrophobic barrier using a barrier pen, and the primary antibody (p16) was applied at 1:100 concentration and diluted in a blocking solution overnight. The next day, slides were rinsed 3 times with PBS 1×, and the secondary antibody was applied at 1:400 concentration diluted in blocking solution for 2 h. Again, slides were rinsed 3 times with PBS 1×, and DAPI staining (1 µg/ml) was applied for 10′. Finally, samples were mounted with coverslips using Vectashield Mounting Medium (Vector Laboratories, Burlingame, CA, USA, H1000). Presence of positive p16 cells were quantified.
Cell culture
Primary human fibroblasts (Thermo fisher; C0135C) were cultured in high glucose DMEM (Dulbecco’s modified media) (Gibco, Invitrogen, Eugene, OR, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Invitrogen, Eugene, OR, USA) and antibiotics (Sigma Chemical Co., St. Louis, MO, USA). Cells were incubated at 37 °C in a 5% CO2 atmosphere. Senescent cells were generated by X-ray irradiation. 6300 cells/cm2 were seeded 24 h prior to 20 Gy irradiation and were used for experiments 7 days later.
Conditioned medium
Irradiated (20 Gy) or non-irradiated cells (2 × 106) were seeded in a 10 cm dish and incubated for 7 days in DMEM with 0.5% FBS. After incubation, the conditioned medium (CM) was collected, centrifuged at 5000 g and filtered through a 0:2 µm pore filter. CM was mixed with DMEM 40% FBS in a proportion of 3–1 to generate CM containing 10% FBS.
Immunofluorescence assay
Fibroblasts were grown on 1 mm width glass coverslips for 72 h in high glucose DMEM medium containing 10% FBS and 1% antibiotics. They were washed twice with PBS, fixed in 3.8% paraformaldehyde for 15′ at room temperature, permeabilized with 0.1% Triton X-100 in PBS for 10′ and incubated in blocking buffer (BSA 1%, 0.05% Triton X-100 in PBS) for 30′. In the meantime, the primary antibody was diluted 1:100 in antibody buffer (0.5% BSA, 0.05% Triton X-100 in PBS). Fibroblasts were incubated overnight at 4 °C with the primary antibody and subsequently washed twice with PBS. The secondary antibody was similarly diluted 1:400 in antibody buffer, but their incubation time on cells was reduced to 2 h at room temperature. Coverslips were then washed twice with PBS, incubated for 5′ with PBS containing DAPI 1 µg/ml and washed again with PBS. Next, they were mounted on microscope slides using Vectashield Mounting Medium (Vector Laboratories, Burlingame, CA, USA, H1000).
SA-β-galactosidase assay and immunofluorescence
5 × 104 IMR90 cells per well were seeded in 6-well plates. Four days later, cells were fixed with 0.5% glutaraldehyde (Sigma) in PBS for 10 min. Fixed cells were washed three times with PBS 1 mM MgCl2 pH 5.7, before adding to each well 2 ml of pre-warmed X-Gal staining solution (2 mM MgCl2, 5 mM K4Fe(CN)6—3H2O, 5 mM K3Fe(CN)6, 1 mg/ml X-Gal solution ready to use (R0941, ThermoFisher) in PBS). Plates were incubated for 2–24 h at 37 °C, washed and imaged. SA-β-Gal activity positive and negative cells were quantified using FIJI/ImageJ.
Healthy control, from BioChain (T1234149), and fresh patient liver tissues were fixated with Aceton for 10 min (4℃). Slides were rinsed 3 times with PBS 1× and blocking solution (2% BSA, 0.05% triton X-100 in PBS 1×) was applied for 1 h. Samples were surrounded by a hydrophobic barrier using a barrier pen. Primary antibody (Anti-beta Galactosidase (ab9361) and Anti-Ki67 (ab15580) antibodies, Abcam) were applied in blocking solution overnight (4℃). The next day, slides were rinsed 3 times with PBS 1×. Secondary antibody was applied in blocking solution for 2 h (RT). Again, slides were rinsed 3 times with PBS 1× and DAPI staining solution (1 µg/ml) was applied for 10 min. Samples were mounted on coverslips in using Prolong® Gold Anti Fade Reagent.
ELISA (enzyme-linked immunosorbent assay)
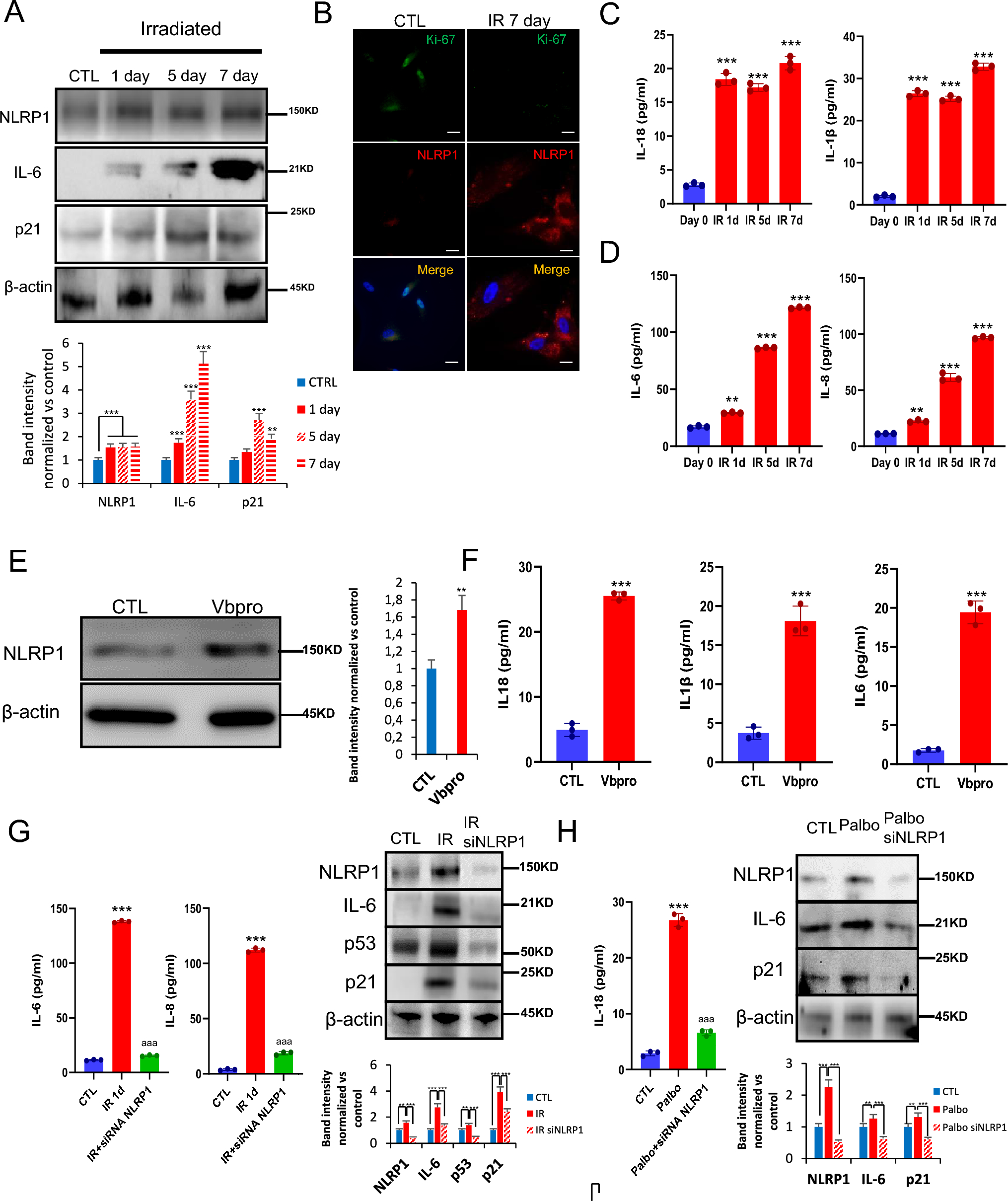
IL-6, IL-8, IL-1β and IL-18 levels were assayed in supernatant by duplicate using commercial ELISA kits (Thermo Fisher Scientific, MA, USA).
Cytokine array
Blood serum was collected from wild‐type and Nlrp1, Nlrp3 and Gsdmd−/− mice. In an in vitro model of senescence induced by X‐ray irradiation (10 Gy) (with or without irradiation), cells were cultured in serum‐free media for 24 h and media were collected for analysis. Media and blood serum were analyzed for expression of several mouse cytokine and chemokines (MD44) or human cytokine and chemokines (HD48), respectively, using a Multiplexing LASER Bead Assay (Eve Technologies).
siRNA transfection
Cells were seeded on 6-well plates until 75% confluence in 2 ml DMEM high glucose medium (Cat. 10566016) supplemented with 10% FBS and 1% antibiotics. Transfection was performed according to the lipofectamine RNAiMAX reagent (Cat. 13778–075) protocol. Briefly, the siRNA-lipid complex was prepared in DMEM medium with 3% lipofectamine and 30 pmol of the correspondent siRNA, and incubated for 5 min at RT to form the silencing complex. Then, 250 µl of the siRNA-lipid complex were added to each well. After 72 h, cells were treated and analyzed for the different conditions. Every reagent, including DMEM medium, was purchased from ThermoFisher (Waltham, MA, USA).
DNA treatment
For DNA extraction, cells were seeded on T75 flasks until 80% confluence, then cells were irradiated (20 Gy irradiation and were used for experiments 7 days later) using X-RAD225, 225 kV. After 1 week, irradiated and control cells were scrapped off and centrifuged at 1.000 g for 5′. DNA extraction was performed using 500 μl lysis buffer containing 100 mM Tris HCl, 100 mM EDTA, 100 mM NaCl, SDS 1%, pH 7.5. Samples were incubated at 65 °C for 30′, and 500 μl Phenol/Chloroform/Isoamyl Alcohol 25:24:1 was added. Samples were mixed by simple inversion and centrifuged at 5.000 g for 5′. 300 μl DNA-containing top aqueous phase was retrieved. For DNA precipitation, samples were mixed with 200 μl 5 M potassium acetate and 400 μl isopropanol. After 12.000 g for 15′ centrifugation, the pellet was washed with 70% ethanol twice and dried for 1 h. DNA pellet was resuspended in TE buffer (10 mM Tris HCl, 1 mM EDTA, pH 7.5) and quantified using a NanoDrop™ One/One from Thermo Fisher (Waltham, MA, USA).
Non-irradiated and irradiated DNA-treated cells were seeded on 6-well plates until 90% confluence in 2 ml DMEM high glucose medium supplemented with 10% FBS and 1% antibiotics. Then, 1 µg/ml of control/irradiated DNA was added for 8 h/24 h. Finally, cells were scrapped off and pelleted for further analysis.
Poly(dA-dT) DNA was transfected using Lipofectamine 2000 at 1 µg/ml concentration for 24 h. Cells were scrapped off and pelleted for further analysis.
Immunoblotting
Western blotting was performed using standard methods. After protein transfer, the membrane was incubated with various primary antibodies diluted 1:1000; the corresponding secondary antibodies were coupled to horseradish peroxidase at a 1:10,000 dilution. Specific protein complexes were identified using the Immun Star HRP substrate kit (Biorad Laboratories Inc., Hercules, CA, USA).
Bioinformatics analysis
We used ARCHS4 [12] to download the samples of the two datasets analyzed referring to mutations in NLRP1 and a model of oncogene-induced senescence in keratinocytes, GSE85791 & GSE180361, respectively. ARCHS4 provides RNAseq data that are uniformly processed using the Kallisto aligner [13]. Filtering and normalization were performed using the TMM method in EdgeR (3.40.1). Differentially expressed genes were obtained using the VOOM function from the Limma package (3.54.0). An FDR < 0.05 was used as a cut-off. Finally, differentially expressed genes from both datasets were individually selected for Gene Set Enrichment Analysis (GSEA) using GSEA from the GSEABase package (1.6.0), selecting hs_gsea_c2 as the geneset.
Statistics
All data are expressed as means ± SEM. After evaluation of normality using Shapiro–Wilk test, statistical differences among the different groups were measured using either an unpaired Student t test or 1-way analysis of variance (ANOVA) when appropriate with Tukeys post-hoc test. A P value of ≤0.05 was considered statistically significant. Statistical analyses were performed using Prism software version 5.0a (GraphPad, San Diego, CA). Asterisks in the figures represent the following: * P ≤ 0.05; ** P ≤ 0.01; and *** P ≤ 0.001.



留言 (0)