The clinical manifestations of chromosome 18q deletion syndrome include psychomotor developmental delays (96%), hypotonia (89%), convulsions (22%), short stature (55%), hypoplasia of the midsection of the face (78%), strabismus (30%), ear anomaly (67%), hearing impairment (72%), aural atresia (4%), cleft lip and palate (67%), microcephaly (59%), limb skeletal deformities (85%), reproductive organ defects (hypoplasia, cryptorchid testes, micropenis) (30%), urinary tract malformation (19%), congenital heart diseases (48%), atopic dermatitis (22%), and hypoventilation [2, 3]. Patients with 18q deletion syndrome exhibit a deficiency in at least one of the immunoglobulins, IgA, M, G, E, and IgG1 − 4 in 88% of cases [2], with IgE deficiency being the most common (52%), followed by deficiencies in the IgG subclass (42%), IgM (40%), IgG (32%), and IgA (20%) [2]. Therefore, 18q deletion syndrome is recognized as a cause of hypogammaglobulinemia [2, 4, 5].
Table 2 presents the clinical features and laboratory findings of the two patients. Patient 1 presented with psychomotor developmental delays, hypotonia, short stature, hypoplasia of the midsection of the face, strabismus, ear anomalies, aural atresia, and cryptorchidism as diagnostic features. Patient 2 exhibited psychomotor developmental delays, ear anomalies, cleft lip and palate, and skeletal deformities of the limbs as diagnostic features. Approximately 94% of 18q deletion syndrome cases are de novo [3], and similarly, both cases in this study were de novo. The patients in the present study exhibited low levels of IgG, IgM, IgA, and IgE. Additionally, patients with this syndrome present with recurrent infections of the respiratory (37%), urinary (19%), and gastrointestinal (19%) tracts, as well as sepsis (11%), indicating signs of humoral immunodeficiency [2, 4, 5]. Patients with 18q deletions frequently experience autoimmune diseases, repetitive infections, and allergies due to immune dysregulation, and present with variable antibody and regulatory T cell deficiencies. Both patients were negative for HIV antibodies and not receiving any drugs for treatment, including antiepileptic drugs. Therefore, secondary immunodeficiency was unlikely.
Table 2 Clinical features and laboratory findingsCVID is associated with a differentiation failure of memory B and plasma cells. CVID is the most common primary immunodeficiency in adulthood [6, 7]. LOCID is a type of CVID that is distinguished from CVID by the presence of T-cell abnormalities. CVID can be distinguished from combined immune deficiency according to TREC and KREC levels [8]; patients with LOCID have low TREC and KREC levels. This classification, based on TREC/KREC levels, correlates well with complications, neoplasms, autoimmune diseases, and opportunistic infections, which are more common among patients with LOCID [8, 9]. Extremely low TREC and KREC levels were observed in the cases presented in the present study.
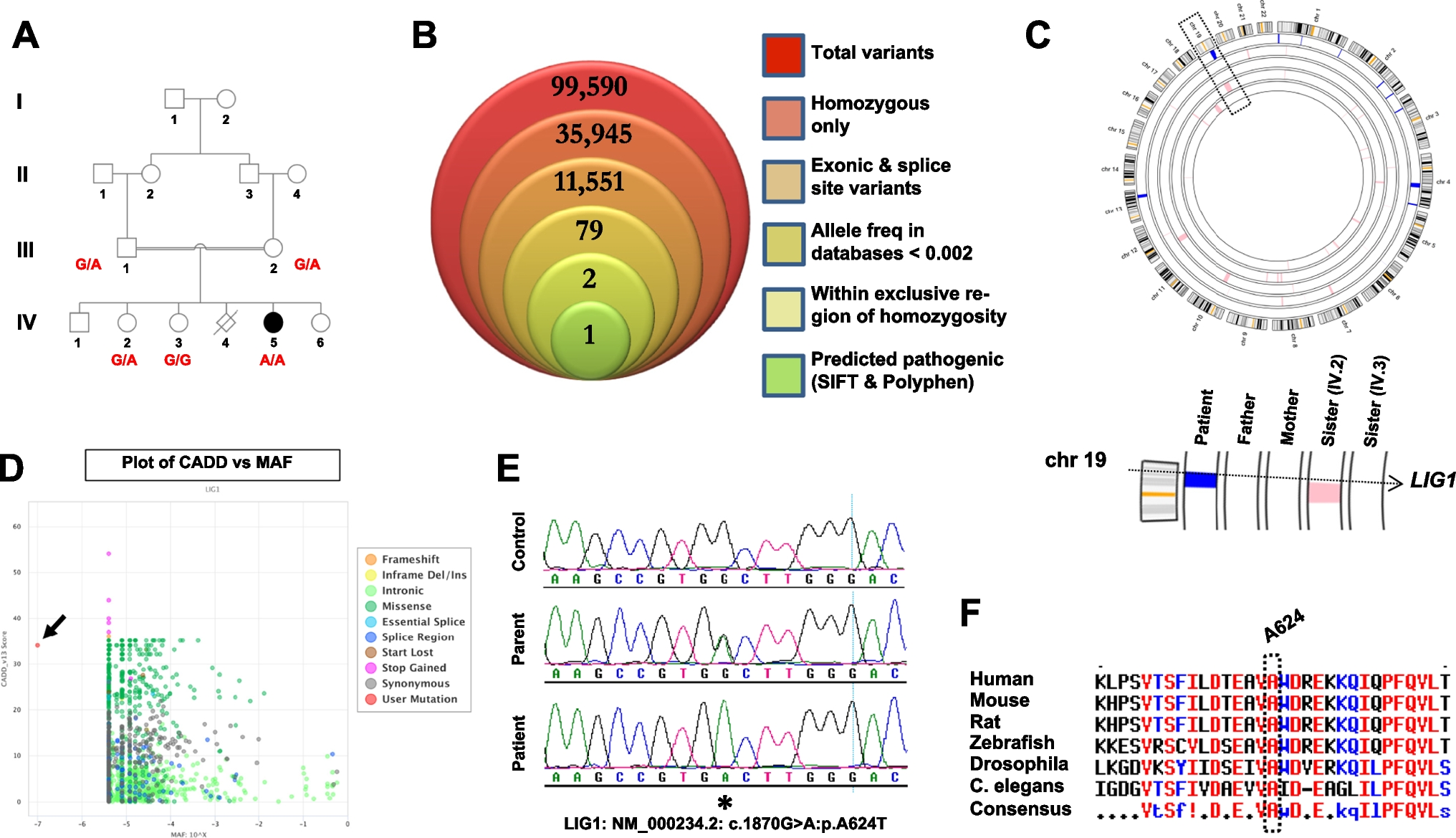
Patient 1 presented with a loss of 18q21.32-q22.3 on the CGH array, whereas patient 2 presented with a loss of 18q21.33-qter. In most patients with 18q deletion syndrome, the deletions are terminal and are localized in the distal half of the long arm (18q21.1-qter) [10], as observed for the patients in this study. Some genes (NEDD4L, MALT1, TNFRSF11A, BCL2, CD226, SOCS6, and NFATC1 in patient 1, and BCL2, CD226, SOCS6, and NFATC1 in patient 2) (Supplementary Tables 1, 2) that may be involved in IEI, were present at the chromosomal deletion sites of the patients. MALT1 has an intrinsic T-cell role in regulating homeostasis; patients with homozygous missense variants in MALT1 have severely impaired T-cell proliferation in response to antigens and antibodies, resulting in a combined immunodeficiency [11]. However, whole-exome analyses showed that patient 1 did not exhibit any morbid variants in the normal alleles (without the deletion of 18q). TNFRSF11A is essential for the correct differentiation of medullary thymic epithelial cells. Moreover, haploinsufficiency of TNFRSF11A and NEDD4L leads to a decrease in regulatory T cells [2, 12, 13]. BCL2 is involved in immunoglobulin synthesis (variants that could cause CVID) and lymphocyte development. CD226 is a signaling molecule that induces cytotoxic activity in T and NK cells. SOCS6 is involved in lymphocyte maturation, and NFATC1 is involved in maintaining regulatory T cell functions [2, 14, 15]. However, none of these genes act in an autosomal-dominant manner. There may be some unidentified genes that correlate with LOCID at sites where 18q is deleted or at other sites with deletions. Although there are many cases involving deletion of the same region as that of the two patients presented in this study, there are no reports of patients with 18q deletion syndrome developing LOCID. It is possible that these patients simply have not yet developed LOCID or that they might not have been adequately assessed for it. Patient 1 had a 21q + abnormality observed through G-banding. DSCR1 is a gene suggested to contribute to the immunodeficiency observed in a proportion of patients with trisomy 21 [16]. The results of the CGH + SNP microarray for chromosome 21 of patient 1 showed deletion of 21q22.3 (Supplementary Fig. 1) and no DSCR1 gene in the deleted region.
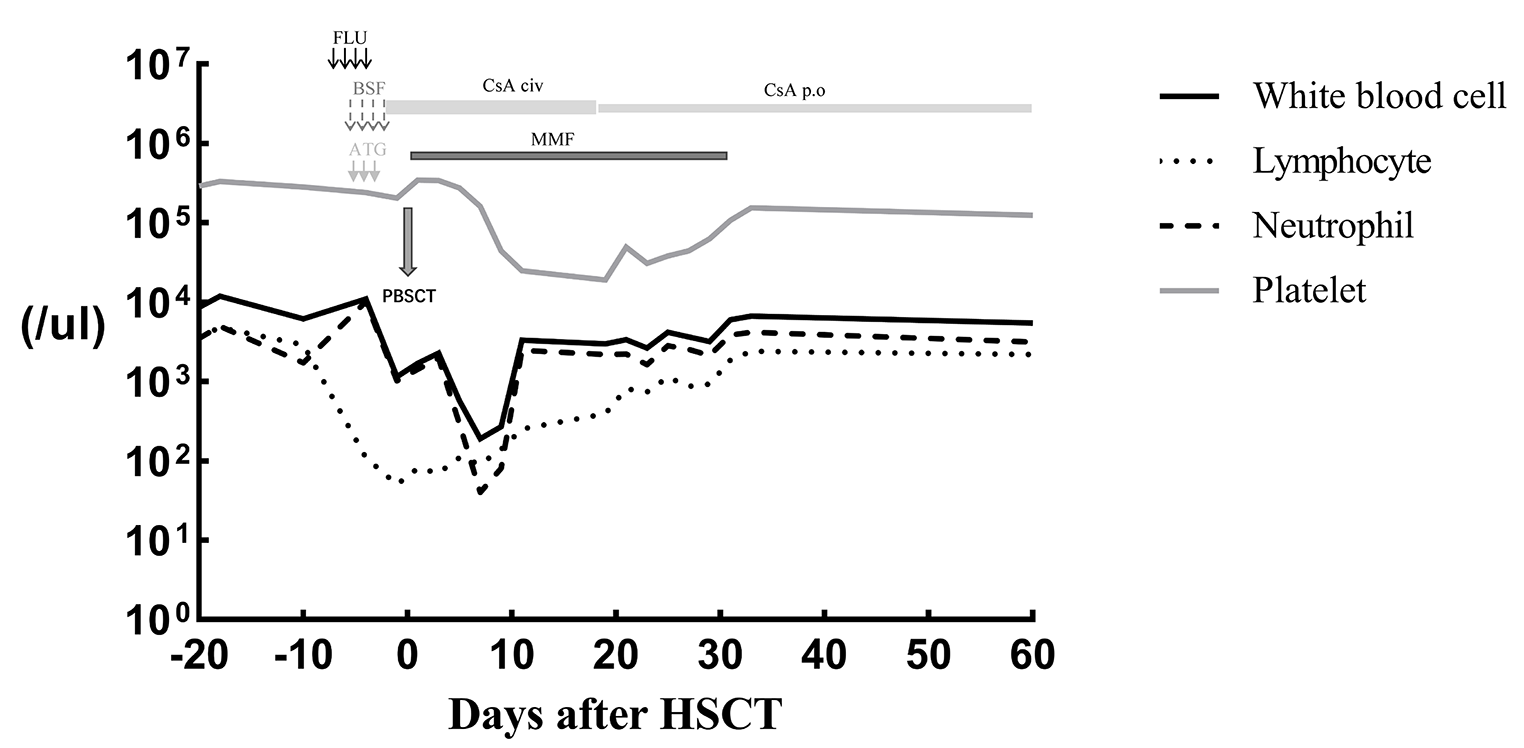
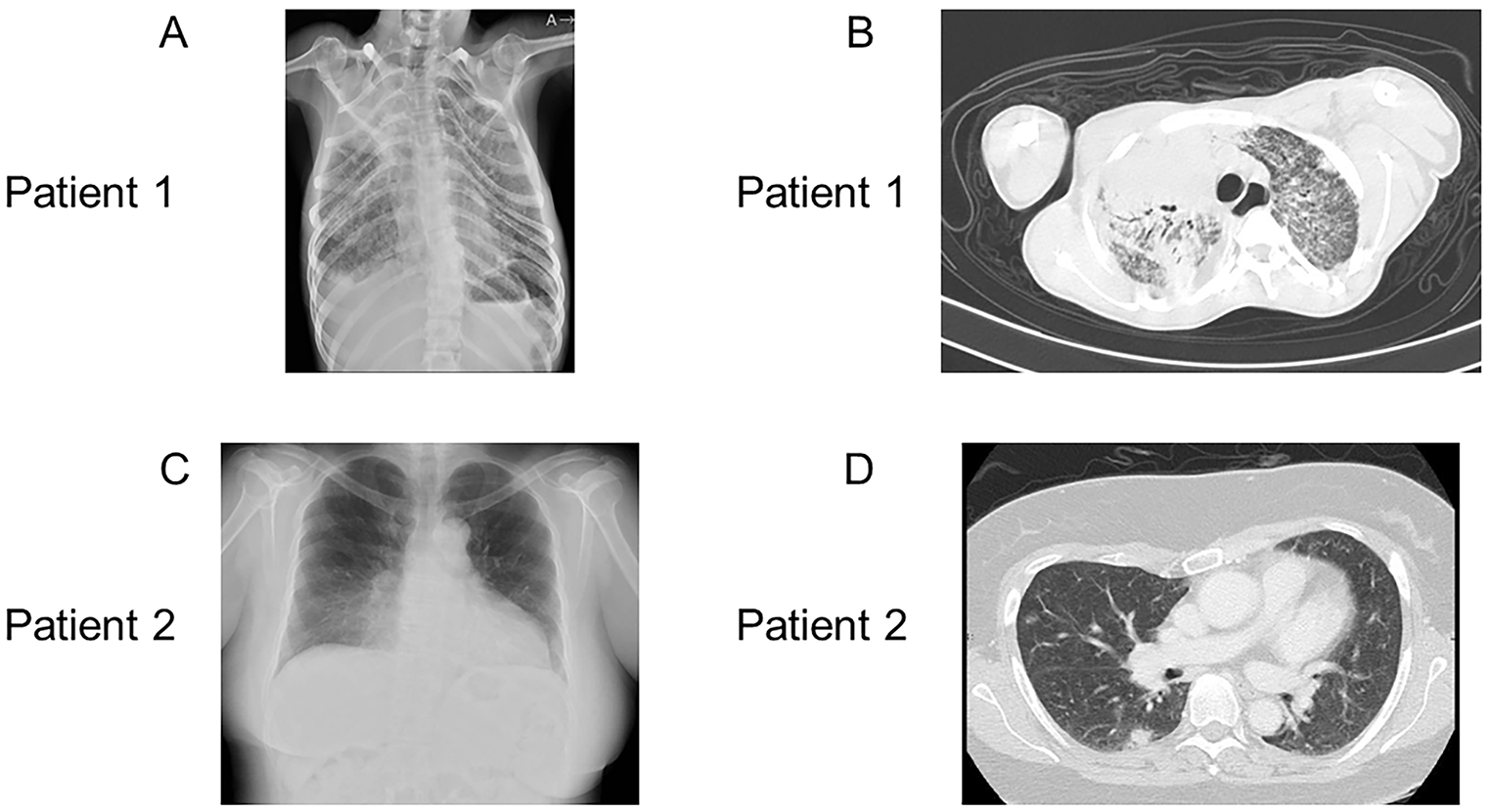
In IEI, susceptible infections and pathogens are characterized according to the type of immunodeficiency, whereas the putative primary immune deficiencies differ according to the type of infection [17]. Patients with antibody deficiency are susceptible to recurrent bacterial respiratory infections, whereas patients with cell-mediated immunodeficiencies are susceptible to opportunistic infections such as PCP. The drug combination ST is required to prevent PCP in patients with cell-mediated immunodeficiency; therefore, this drug combination is also administered orally to patients after a confirmed diagnosis. Periodic substitution of immunoglobulins is used to treat patients with hypogammaglobulinemia. Although adaptations for allogeneic hematopoietic cell transplantation could be considered for patients with LOCID, Patient 1 is believed to have had difficulty adapting to transplantation because of his general condition. Patient 2 would be considered for transplantation during the follow-up.
Patient 2 had not been diagnosed with the 18q deletion syndrome earlier. 18q deletion syndrome is associated with characteristic malformations; therefore, it should have been suspected, and chromosome testing should have been performed. Patient 1 was diagnosed with 18q deletion syndrome earlier but was not tested for immunocompetence. The occurrence of these two cases suggests that patients with 18q deletion syndrome should be tested for cellular/humoral immunocompetence regularly, at least once a year. When encountering patients with PCP, immunocompetent workups should be conducted, and attention should be paid to the fact that LOCID, the most severe immunodeficiency, can affect patients with 18q deletion syndrome. Although patients with 18q deletion syndrome are known to develop CVID, there is no recognition that they might develop LOCID; therefore, physicians should be aware of this aspect. The immune evaluation of these patients had several limitations. HIV antigen/PCR test was not performed, dihydrorhodamine 123 test was not performed for patient 1, antibody responses to pneumococcal serotypes were not assessed, and T-cell proliferation assays were performed only once.
In conclusion, to the best of our knowledge, we report the first diagnoses of two patients with chromosome 18q deletion syndrome who presented with LOCID. Our study highlights that patients with 18q deletion syndrome who present with recurrent or severe infections should be regularly tested for cellular/humoral immunocompetence.



留言 (0)