
記住我


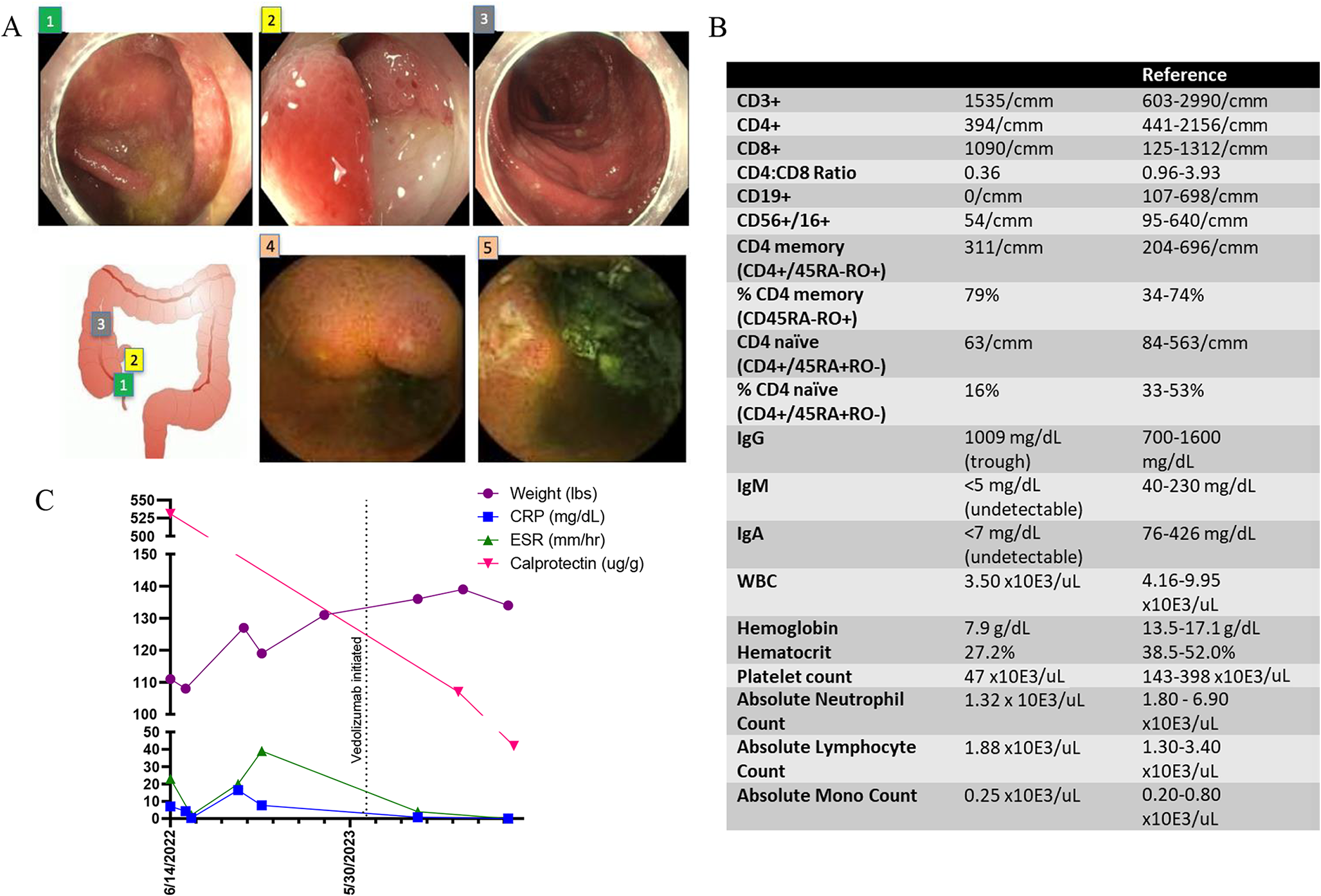
The patient is a product of 32 week gestation, delivered by Cesarean section due to intrauterine growth retardation (IUGR), with a birth weight of 2.3 kg. She was admitted to a local hospital at the age of 2 and 3 months with recurrent chest infections and anemia. At 4 months, the patient developed perianal abscess associated with neutropenia and localized BCGitis. Wound culture grew Proteus mirabilis and Citrobacter freundii. The parents are 1st degree cousins, and the patient has one healthy brother and three healthy sisters, with a history of previous neonatal death (a preterm 28 week newborn with multiple congenital anomalies and non-immune hydrops), but no family history suggestive of immunodeficiency (Fig. 1A).
Fig. 1
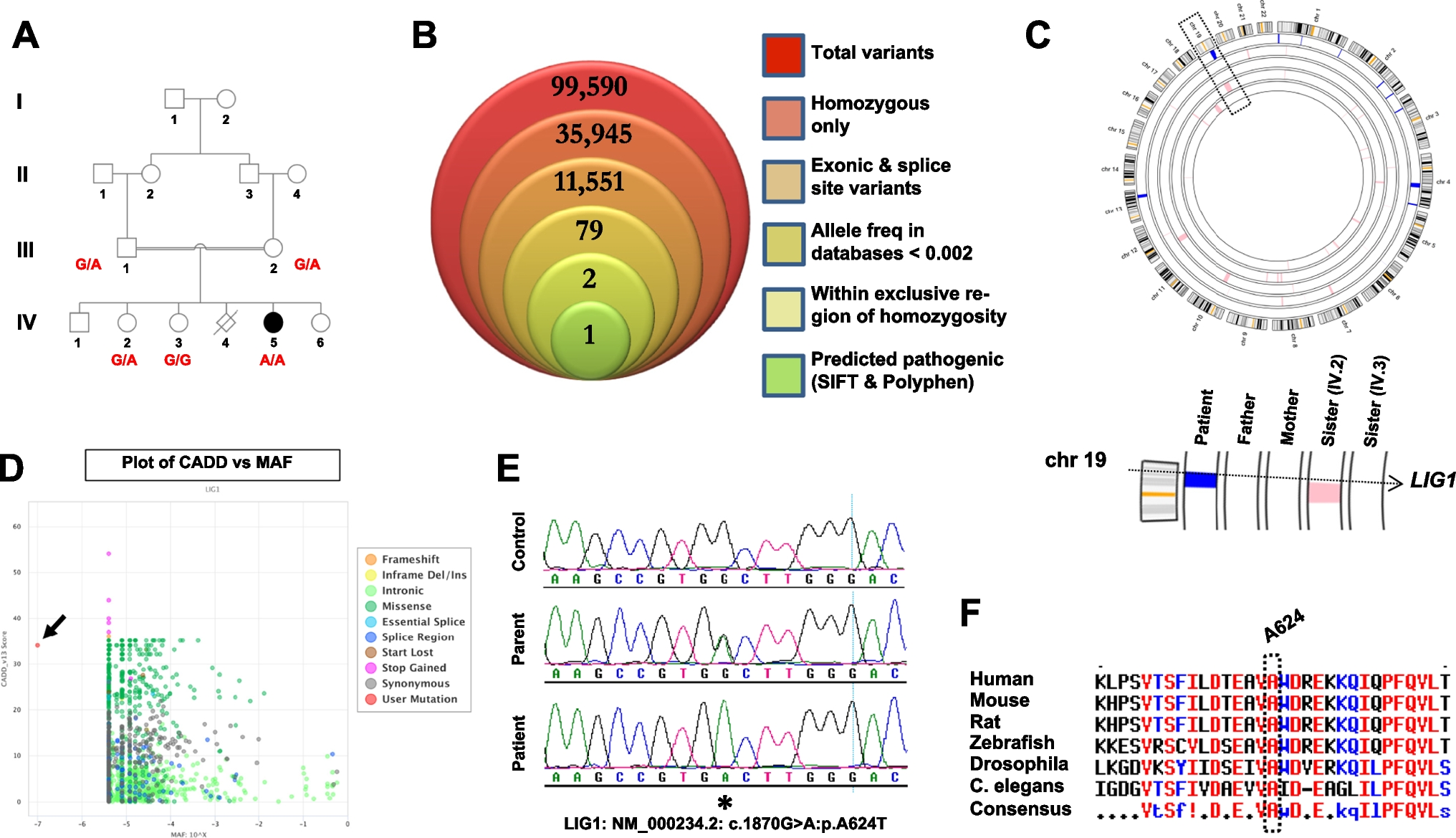
A novel LIG1 mutation is linked to a severe combined immunodeficiency phenotype. A Pedigree of the family in this study, along with genotypes for the individuals who were available for recruitment. B WES filtering scheme, shown as a stacked Venn diagram illustrating the total number of variants observed following the addition of each filter. C Output from AgileMultiIdeogram, with all regions of homozygosity colored blue (patient) or pink (all other family members) across the genome. Bottom portion shows a close-up of chromosome 19 and the LIG1 locus. D PopViz2 output (shiva.rockefeller.edu/PopViz2/) plotting CADD scores of all known LIG1 variants against their associated minor allele frequencies. Arrow indicates the A624T mutation. E Sequence chromatogram from the patient, alongside a parent and a healthy control for comparison. Asterisk denotes the site of mutation. F Protein sequence alignment indicates that A624 is universally conserved across species down to worm
The patient was admitted to the hospital at 5 months of age. She was underweight (3.95 kg) and microcephalic with head circumference of 37 cm. She had indurated BCG site with multiple discoloration of the skin. Respiratory examination showed equal bilateral air entry with transmitted sound. Abdomen was soft and lax with no organomegaly.
Immunological work up showed leukopenia, neutropenia and lymphopenia, with counts of 4.87 × 109/L, 0.39 × 109/L, and 1.61 × 109/L, respectively. She had macrocytic anemia, with a hemoglobin of 81 g/L with normal platelets count. She had pan-hypogammaglobulinemia with 0.83 g/L of IgG, less than 0.5 g/L of IgA, and less than 0.25 g/L of IgM. Lymphocyte subsets showed CD3+ of 310 /mm3, CD4+ of 129 /mm3, CD8+ of 51 /mm3, CD19+ of 342 /mm3, and CD16+CD56+ of 265 /mm3. Percentages of CD3+CD45RA+ and CD4+CD45RA+ were 19% and 4%, respectively. Lymphocyte stimulation to phytohaemagglutinin (PHA) was severely depressed (7612 CPM, 6% RR to control).
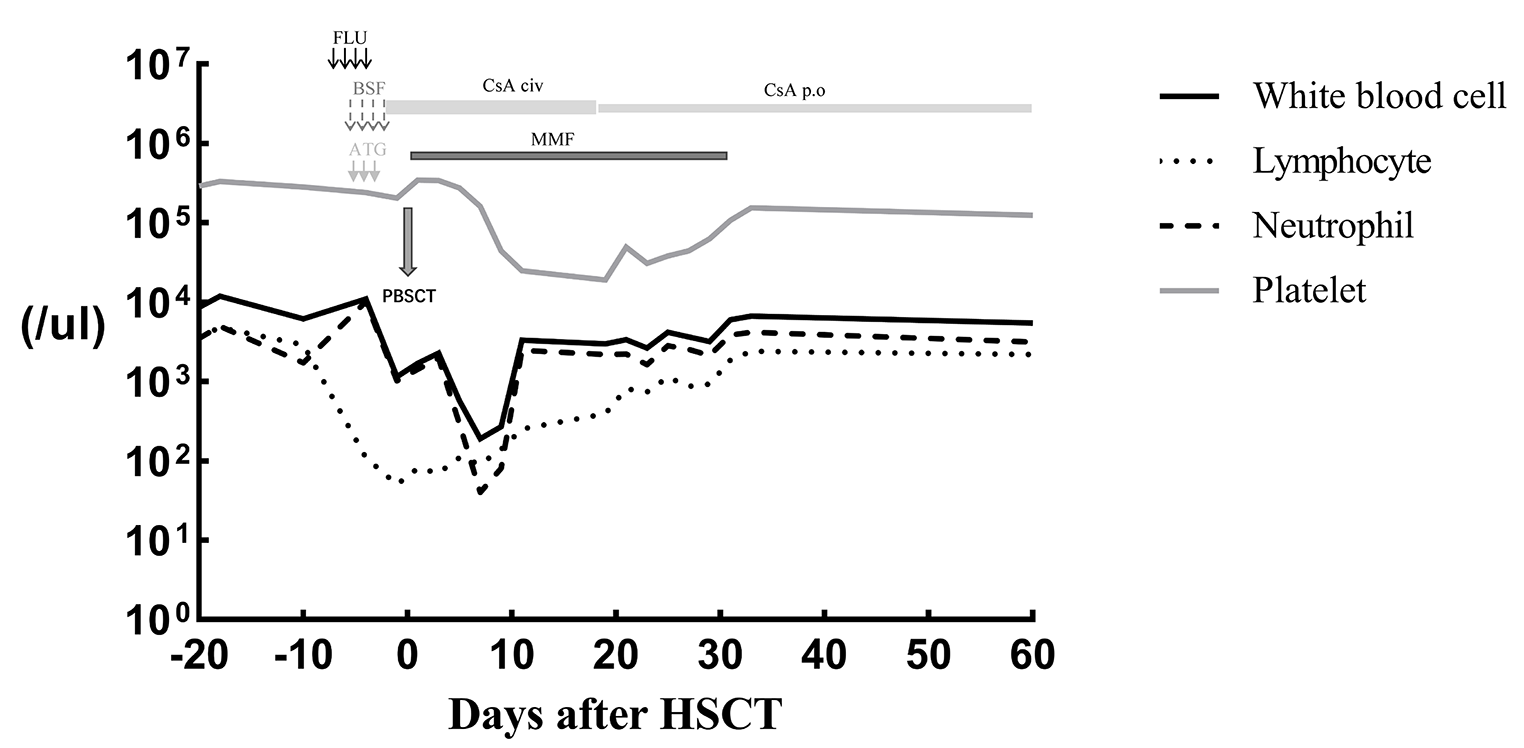
The patient received hematopoietic stem cell transplantation (HSCT) from her HLA-matched sister without conditioning at the age of 6 months. Her BCG infection was treated with anti TB therapy. She is currently doing well 6 years post-HSCT with 87% lymphoid and 4% myeloid engraftment and with no recurrent infections. Her weight remains low (beneath the 3rd centile). She is maintained on weekly subcutaneous immunoglobulin therapy.
Table 1 provides a summary of the clinical, immunological, and genetic characteristics, as well as HSCT outcomes, of all documented LIG1 cases including the present study.
Table 1 Clinical and immunological characteristics, and patient outcomes, of all reported LIG1 deficienciesIdentification of a Novel LIG1 MutationThis family was recruited under an institutional review board-approved protocol, as part of our ongoing effort to identify the genetic causes of rare immunodeficiencies in Saudi Arabia. Due to the consanguineous nature of this family we posited that the underlying mutation was likely present within an autozygous block in the patient’s genome, which would not be shared by any of the other family members. We therefore performed whole exome sequencing (WES) on the patient’s DNA, along with a cataloging of genome-wide genotypes for all family members we had access to (Fig. 1A).
Variants obtained from the WES data were filtered as shown in Fig. 1B, then cross-referenced with the location of all autozygous intervals that were exclusive to the patient (Fig. 1C). This resulted in two variants. One of these was located in INTS11, the catalytic subunit of the Integrator complex which is involved in the 3’ processing of small RNA molecules (NM_017871.6:c.1445C > T:p.T482I). Through the use of SIFT and Polyphen-2, two online prediction programs, we eliminated this variant from consideration as both algorithms predicted this change to be non-pathogenic (SIFT: 0.78, tolerated; Polyphen-2: 0.001, benign).
This left us with a single variant, a missense mutation in the LIG1 gene (NM_000234.2:c.1870G > A:p.A624T) with high pathogenicity prediction scores (SIFT: 0.01, affect protein function; Polyphen-2: 1.0, probably damaging). Plotting its predicted pathogenicity using a third tool, CADD (Combined Annotation Dependent Depletion), revealed a score which was only slightly below that of truncating mutations (Fig. 1D). Sanger sequencing confirmed the mutation and verified its segregation in the family according to the disease state (Fig. 1A, E), while the genotyping data confirmed that the mutation was located within an autozygous stretch that was exclusive to the patient and not shared with the other family members (Fig. 1C, and Fig S1 in Supplementary Material). Protein sequence alignment indicated that the Alanine-624 residue is universally conserved across species down to zebrafish, drosophila and worm (Fig. 1F).
Immunophenotyping of Patient CellsTo functionally characterize the immunological effects of the LIG1 mutation, peripheral blood mononuclear cells (PBMCs) from patient and controls were stained with fluorescent cell-surface antibodies followed by flow cytometric analysis. There was a drastic reduction of CD3+, CD4+ and CD8+ T-cell subtypes, as compared with pediatric and adult controls (Fig. 2). The readings from these PBMCs, sampled at the age of 6 months, are far below the age-matched 10th percentile measurements that have been published for healthy cohorts [15, 16].
Fig. 2
Immunophenotyping of patient PBMCs versus pediatric and adult controls. A Investigation of T, B and NK fractions reveals depressed levels of CD3+ T cells in the patient, including helper and cytotoxic subsets. B Analysis of CD3+CD4+ and CD3+CD8+ compartments showing percentages of naïve (CD45RO−/CD27+), central memory (CD45RO+/CD27+), effector (CD45RO−/CD27−) and effector memory (CD45RO+/CD27−) subsets, as well as regulatory T cells (CD4+CD25hiCD127lo). C Assessment of CD19+ B cells indicating percentages of naïve (CD27−/IgD+) and class-switched memory (CD27+/IgD−) subsets, as well as transitional (CD38hi/CD24hi) and plasmablast (CD38+/CD24−) fractions
Specific analysis of helper T cells (CD3+CD4+) showed that < 40% of these were naïve. This is considerably below Schatorje and colleagues’ lower limit for this age group. Conversely 18% of CD4+ T cells exhibited an effector memory phenotype, whereas the highest observed value in healthy age-matched (5–9 month old) babies was reported at only 1% (Fig. 2) [15]. Our patient’s cytotoxic T cell effector memory percentage was, likewise, well outside the range of our pediatric controls. The central memory helper T subset value lies outside our controls but is within the range provided for our patient's age at sampling.
As with T cells, percent mismatches were also observed with the CD19+ B cell population. The proportion of naïve patient B cells was at the top range of our controls. The transitional B cell subset was markedly higher than controls, while the percentage of class-switched memory cells was drastically lower (Fig. 2).
Patient Cells Reveal Reduced Proliferation and Viability, and Increased RadiosensitivityTo study the impact of the mutant LIG1 gene at the cellular level, skin biopsy was taken from the patient following her guardian’s informed consent. Fibroblast cells derived from the biopsy were noted to have a significantly slower growth rate than controls, and this was confirmed using crystal violet staining across different seeding densities and timepoints (Fig. 3A). We also checked the effect of H2O2 on cell recovery by comparing crystal violet (biomass) readout of cells without treatment, to the readout following 3 days of recovery post-treatment. Data indicated a poorer recovery rate for the patient cells for some durations of treatment, but not for others (Fig. 3B). Additionally, flow cytometry analysis revealed a significantly higher percentage of apoptotic cells in patient fibroblasts as compared to controls, as determined by DAPI staining. This trend was clear in the absence of DNA damage induction, and was exacerbated in its presence (Fig. 3C).
Fig. 3
Patient fibroblast cells show diminished proliferation, survival, and viability. A Patient and control fibroblast cell lines were seeded at low or high densities, then harvested at various timepoints and analyzed using crystal violet as a proxy for determining growth rates. B Cells were tested for their ability to recover following treatment with 200 µM H2O2 for the indicated time periods, as outlined in the Methods section. Crystal violet readings from day 3 (post-recovery) were normalized to the corresponding day 0 readout. C Viability (the DAPI-negative percentage) of patient and control fibroblast cell lines was determined using flow cytometer, in the presence or absence of 200 µM H2O2 treatment for 30 min. For all experiments a total of 3 control cell lines were utilized. Data are based on three independent experiments for the proliferation and viability assessments, and four for the survival assay. Asterisks denote p-value significance (*p < 0.05, **p < 0.01, ***p < 0.001; unpaired Student’s t-test) and n.s. denotes not significant
We hypothesized that patient cells would be susceptible to buildup of DNA damage, even in the absence of external agents. By immunoblot analysis we observed that patient fibroblasts did indeed exhibit buildup of γ-H2AX, an established marker of the early cellular response to double-stranded DNA breaks (Fig. 4A) [17]. This finding was reproducible and the difference between patient and controls was significant. We also assessed protein levels of LIG1 and observed a non-significant trend of greater expression in the patient cells (Fig. 4B). To investigate if the missense variant was affecting protein stability, whether positively or negatively, human embryonic HEK293 cells were transfected with FLAG-tagged LIG1 plasmid using both the wildtype (WT) and mutant forms. Immunoblot analysis was used for comparing the exogenous LIG1 protein levels (Fig. 4C). The data did not reveal any notable difference between the two groups, suggesting that pathogenesis was not due to any changes in protein stability.
Fig. 4
LIG1 patient fibroblasts show enhanced susceptibility to DSB damage. A Immunoblot of fibroblast protein lysates, in the absence of any treatment, reveals high basal levels of γ-H2AX staining in the patient. Both beta actin and PCNA were used as loading controls. Image is representative of three independent experiments. B Quantification of the immunoblot data, based on normalization of the γ-H2AX readings to PCNA. Normalization to beta actin revealed the same pattern (data not shown). C Top: immunoblot of cell lysates from HEK293 cells transfected with FLAG-tagged wildtype and mutant LIG1 expression vectors. Bottom: aggregated data (from 12 transfections) reveals no significant difference in the FLAG-to-GAPDH ratio between the two vectors. Error bars indicate SEMs. D Radiation sensitivity assay. The patient and two control fibroblast cell lines were either left untreated or were subjected to 4 Gy ionizing radiation, then allowed to recover for the given time periods. The box plots demonstrate that patient cells had considerably higher γ-H2AX staining at all time periods. For each experiment an average of 45 cells per cell line were scored for every time point, and data are based on two independent experiments. Asterisks indicate significance levels (*p < 0.05, **p < 0.01, ***p < 0.001; unpaired Student’s t-test). Error bars indicate SEMs
Next, we investigated the effect of another DNA damaging agent by conducting a radiosensitivity assay. Fibroblasts were seeded on poly-L-lysine coated coverslips, then exposed to 4 Gy ionizing radiation and allowed to recover for 3, 24 or 48 h, along with a no radiation control. Probing the cells with fluorescently-labelled anti-gamma-H2AX revealed higher baseline staining in the patient cells, in the absence of treatment (Fig. 4D). This was in line with our immunoblot results. Additionally, at all recovery timepoints the patient cells exhibited significantly higher numbers of gamma-H2AX foci per nucleus, indicating more susceptibility to radiation and a slower rate of DNA damage repair. A previous radiosensitivity study on LIG4-deficient fibroblasts indicated that these, on average, generated three times as many gamma-H2AX foci per nucleus as a wildtype line across all timepoints [18], which is largely analogous to our LIG1 findings. By comparison, fibroblasts deficient in Artemis, another SCID-causing gene with a role in DSB repair, generated substantially fewer foci than the LIG4 mutant cells and were faster in returning to basal levels.
Cell Cycle Defects in Patient CellsWe noted that growth rates for the patient fibroblast cells were drastically slower than controls, suggesting a cell cycle defect. To examine cell cycle characteristics in the presence or absence of induced DNA damage, cells were assessed as-is or were exposed to 4 Gy ionizing radiation then allowed to recover for up to three days. At various timepoints cells were treated with propidium iodide and observed by flow cytometry to track their cell cycle. At baseline (without treatment), the majority of patient cells were in G1 (Fig. S2A), at a percentage significantly higher than controls, and with significantly fewer cells in S and G2 (Fig. S2B). This reinforced our earlier observation of impaired patient cell growth and suggested G1/S checkpoint difficulties. Following radiation, patient cells were depleted from G1 and began accumulating in G2 at significant levels, and this indicated that the previous buildup in G1 had not been due to increased senescence (G0). Even 72 h post-radiation, the percentage of cells that had effectively exited G2 was low. This contrasted with control cells which exhibited a G2/M spike at 24 h, but then were largely able to progress through mitosis so that by 72 h the percentage of cells in G2 had reverted to basal levels (Fig. S2B).
A624T Displays Reduced Catalytic ActivityHuman LIG1 ligation catalysis and fidelity are controlled by two Mg2+ binding sites [19]. The first Mg2+ (called catalytic Magnesium) is strictly required for catalysis and provides some basic level of fidelity against a limited number of substrate defects before committing to ligation. The second Mg2+ (called high-fidelity Magnesium) causes the enzyme to terminate ligation on substrate defects. A624 is part of a beta sheet (aa. 616–626) involved in the interaction with the catalytic Mg2+ ion. Additionally, molecular dynamics simulations (see below) suggest that the effects of the A624T mutation also propagate allosterically to residues involved with the high-fidelity Mg2+. These structural considerations prompted us to evaluate the effect of A624T on LIG1 catalytic efficiency and fidelity.
LIG1 activity was monitored using a ligation assay where a synthetic nick substrate is converted to a full dsDNA duplex upon ligation (Fig. 5A). The substrate was phosphorylated at the 5’end of the 5’arm of the nick to allow ligation. The same 5’arm was labeled at the 3’end with Cy5 fluorophore for reaction readout. The terminal 3’end nucleotide of the 3’arm of the substrate was varied to either a perfect-match natural nucleotide (dT), a mismatch (dG) or several chemically modified nucleotides (oxoG, NI, dU, and dI) (see Methods). Ligation fidelity should force LIG1 to abort activity on all substrates except for the perfect-match dT. Nevertheless, it was previously shown [19] that WT LIG1 tolerates 8-Oxoguanine at the 3’end of the 3’arm of the nick when base-paired to a natural deoxyadenosine (dA).
Fig. 5
Reduced catalytic activity and enhanced replication fidelity in the A624T mutant protein. A Schematic representation of the ligation substrate and reaction. The nick substrate consists of three pre-annealed oligonucleotides: a 48nt template, a Cy5-labeled 24nt 5’arm phosphorylated at the 5’end to allow ligation, and a 24nt 3’arm containing either a perfectly-matched deoxyribonucleotide or a damage at the 3’end. B Denaturing Urea-PAGE gels showing the ligation product of nick substrates containing either a perfectly-matched deoxyribonucleotide (dT) or different damages at the 3’end of the 3’arm. C Quantification of ligation product as a percentage of total substrate from the ligation gels showed in panel B. The bar chart illustrates the mean (as bar height) and one standard deviation (as error bar) of three independent measurements. The p-values compare between the ligation generated by LIG1 WT versus A624T and are reported to the most significant digit. D Denaturing Urea-PAGE gels showing the ligation products for a nick substrate containing a perfectly-matched deoxyribonucleotide (dT) at the 3’end of the 3’arm at various Mg2 + concentrations for WT versus A624T mutant. E Quantification of ligation product as a percentage of total substrate from the ligation gels showed in panel D versus Mg2 + concentration. The data points illustrate the mean (as point location) and one standard deviation (as error bar) of three independent measurements. For both WT and A624T, the experimental data points were fitted to Hill equations with a fixed Hill coefficient of 2 to account for the two Mg2 + binding sites present in the enzyme
The first clear insight into the effects of the A624T mutation was offered by monitoring ligation of the cognate substrate that contains a perfect-match (dT paired to template dA) natural nucleotide at the 3’end of the 3’arm of the nick (Fig. 5B, C; dT). On this substrate, ligation by the mutant LIG1 was ~ 1.5-folds lower in yield as compared to WT. Furthermore, LIG1 A624T required higher Mg2+ concentration than LIG1 WT to achieve the same ligation yield in the same amount of reaction time (Fig. 5D). Quantitative characterization showed that LIG1 A624T has an ~ 2.5-folds weaker response to Mg2+ titration than the WT protein (Fig. 5E). Collectively, these results indicate that A624T causes a reduction in the catalytic activity of LIG1, which in turn may increase the genome-wide number of un-ligated nicks and cause genomic instability.
A624T Leads to Enhanced Ligation FidelityThe second insight into the mechanistic effects of the A624T mutation was offered by the investigation of ligation fidelity on sub-optimal substrates (Fig. 5B, C; oxoG, NI, dG, dU, and dI). Substrates containing 5-Nitroindole (NI; universal nucleobase), deoxyInosine (dI; nucleoside), and deoxyGuanosine (dG; nucleobase mismatch) at the 3’end of the 3’arm of the nick were well discriminated by both A624T and LIG1 WT and resulted in an almost complete lack of ligation. The substrate containing deoxyUridine (dU; nucleoside) was better discriminated by A624T as compared to the WT by ~ 1.5-folds. Nevertheless, this difference is within the range of the difference in catalytic activity between A624T and WT, thus it may not reflect a true difference in fidelity. The most striking observation is seen in the case of the substrate containing 8-Oxoguanine (oxoG; lesion) at the 3’end of the 3’arm of the nick. This substrate was ligated by WT with a yield similar to that of the cognate substrate (dT), yet it was almost completely aborted by LIG1 A624T (Fig. 5B). Accounting for the ~ 1.5-folds difference in catalytic efficiency of the two enzyme variants, quantification analysis shows that A624T has > 50-folds improved fidelity against 3’end oxoG as compared to the WT protein.
Molecular Dynamic Simulation Reveals Allosteric Effects in the Mutant ProteinAs a corollary to our substrate studies, we conducted molecular dynamic (MD) simulations to explore how A624T might perturb the protein-substrate complex. Using coarse-grained force field, we identified clear localized differences in three specific protein loops between WT and mutant (335–344, 640–646 and 853–865; Fig. 6A). All these loops were oriented towards the dsDNA. The 640–646 loop is the main adenylation domain, and the 335–344 loop is used by the DNA-binding domain to form the H2O network that stabilizes the high-fidelity (HIFI) Mg [19].
Fig. 6
Effects of the A624T mutation on protein loop dynamics. A Ligase 1 has three domains, the A624T mutation is in adenylation domain (green spherical representation). The conformational display of the protein exhibited during MDs is shown for WT LIG1 and A624T. The superpositions of protein structure dynamics are shown to highlight the three protein loops that exhibited different molecular dynamics in red (WT) and green (A624T). Protein loop 335–344 is directly involved in stabilizing HIFI Mg in the DNA-binding domain, whereas protein loop 640–646 is involved in DNA stabilization. Alpha helices, beta sheets and the remaining protein loops are shown in yellow, blue and gray respectively, and DNA in magenta. B The atomic coordination adjustment (Å) undergone in the vicinity of the A624T mutation is shown. The mutated amino acid is shown by the spherical representation, the rest of the side chain atoms are shown in the stick representation with oxygen and nitrogen atoms in red and blue respectively. C The residue interaction network for WT ligase 1 was created to show the physical contacts connecting the mutation site to the affected protein loops through the LIG1 domains. The 335–344 and 640–644 loops of the protein are highlighted with green ovoids
The relative bulking of the mutant threonine side chain, nearly doubling the size of the wildtype alanine, was shown to add a polar moiety oriented towards the back of an alpha helix, leading to the secondary structure of the protein-DNA interface (Fig. 6B). A624T also affected the packing of the side chains and the backbone of the adenylation domain core in the vicinity. The adjustment needed to avoid atomic clashes appeared to be changing the atoms’ coordination in a range of 0.6 Å-2.4 Å; a range that could alter not only the H-bond network, but also other non-covalent interactions involved in stabilizing protein structure and function.
Interestingly, position 624 is not part of the protein-DNA contact interface, the protein loop that stabilizes DNA binding (640–646), nor is it close to the HIFI Mg-stabilizing region in the DNA binding domain. Therefore, we decided to perform a Residue Interaction Network (RIN) analysis to study the mutation’s allosteric effect. The RIN analysis of WT LIG1 showed no direct connection between position A624 (Fig. 6C) and the 335–344 protein loop, or with E592, both of which are involved in HIFI Mg stabilization. However, after introducing the mutation, the surrounding amino acids readjust to a more stable and favorable configuration that seems to alter the RIN (Fig. 6B).
We next compared the conformational sampling obtained during MDs and RIN analysis to determine which connections were lost or gained during the simulation (Fig. 7A). While the WT kept E592 well connected to the protein loop 335–344 (in charge of HIFI Mg stabilization) via a molecular bridge of S588 and Q591, the mutant favored their disconnection, causing an increase of their molecular dynamics and affecting the range of their displacement towards the protein-dsDNA contact interface (Fig. 6A, in green). This disconnection of the protein loop 335–344 is due to a decrease in the constraints applied by the sub-networks identified by RIN (highlighted in green and magenta, Fig. 7B-D). While the A624T mutation expands its effects to the core of the adenylation domain through amino acids R641 and Y598 towards S588, there appears to be a decrease in π-π interactions over the F587-H577 sub-networks and a loss of Van der Waals interactions and H-bonding at E573 that no longer constrain the orientation of the loop where K541 is located (Fig. 7D). This amino acid (K541) is important for the proper location of Q591 and the proper conformation of E592 towards Mg (Fig. 7C).
Fig. 7
Effects of the A624T mutation on the connectivity network of LIG1. A Comparison of Residue Interaction Network (RIN) analysis of WT and A624T proteins is shown. Connectivity changes are illustrated with red or green letters, for loss and gain of a physical interaction. The residue network connecting the T624 mutation to protein loops 335–344 and 640–646 is highlighted with yellow circles (amino acids in both protein loops are identified with green ovoids). Two supporting networks stabilizing the main connectivity network are presented as magenta and green nodes. B 3D amino acid coordination of the LIG1 WT connectivity network (color-coded as in panel A) is presented. The mutation at 624 is shown as spherical atoms and hydrogen bonds are shown as thin black lines. C Stabilization of Mg (lime green) by E592 is shown, as well as the involvement of the supporting sub-network K541, E456 and F507 (dark green). Also shown is the stabilization of Q591 by H-bonds that allow the orientation of E592 toward the Mg HIFI atom. D The effect of the A624T mutation on E592 and HIFI Mg seems to be related also to the loss of π-π stacking interactions (F587 and H577), H-bonds and Van Der Walls connections (R573) that stabilize the protein loop where K541 is located. This therefore does not allow the stabilization of E592 through its backbone connection with Q591
The MD results are in agreement with previous reports, where alanine scanning revealed that alteration of E346 (HIFI Mg stabilization section of the DNA binding domain) or E592 could reduce the kinetic constants of LIG1 by up to three times [19]. A series of rapid quenching experiments showed that WT and a E346A/E592A double mutant exhibited similar performance activities with a normal DNA substrate, proving that these two mutations did not disrupt catalysis. Collectively, our data suggest an allosteric effect of the A624T mutation on the protein loops involved with HIFI Mg as well as the adenylation domain’s DNA binding (loop 640–646).
留言 (0)