
記住我


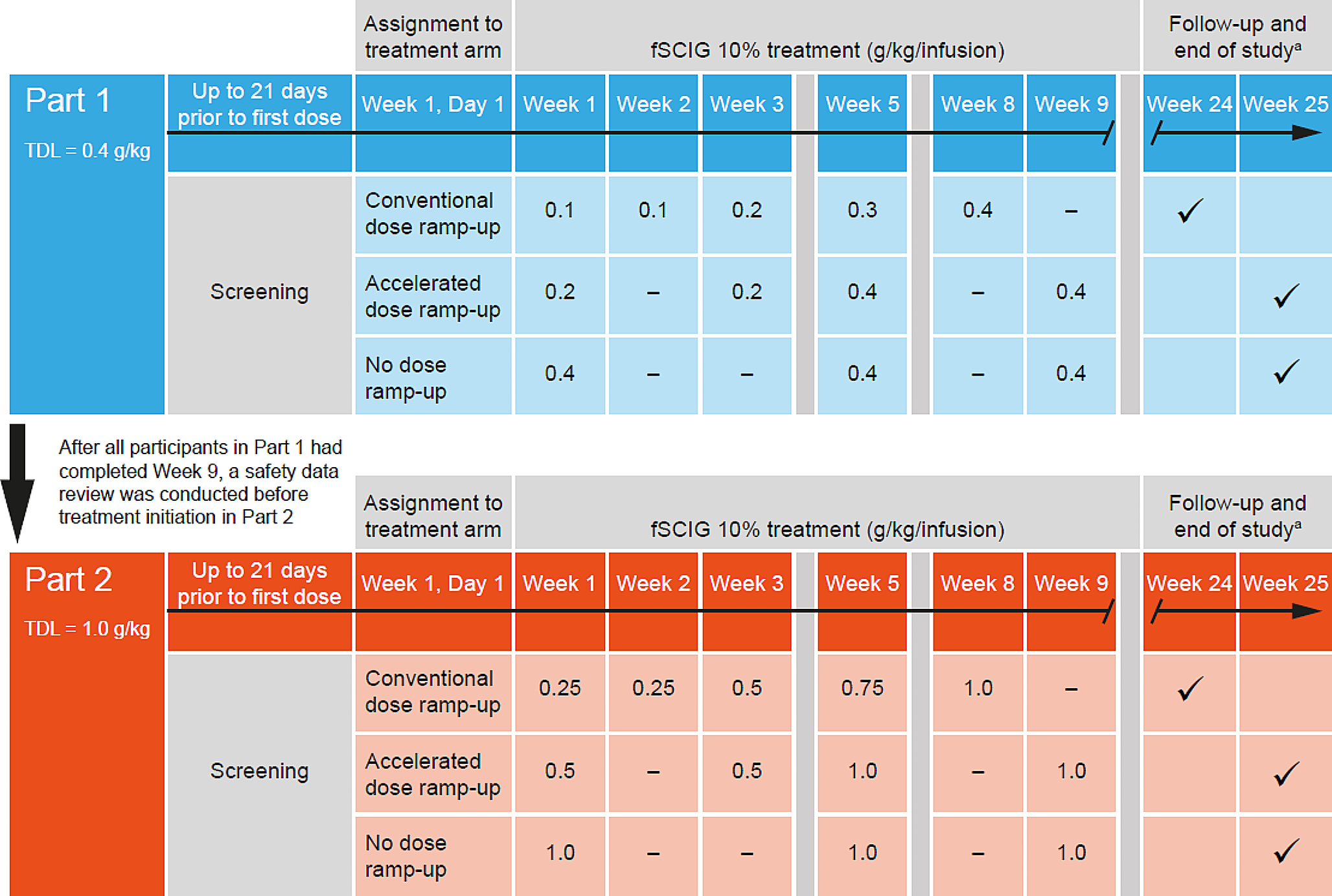
This was a phase 1, open-label, single-center study (NCT04578535) of fSCIG 10% (HYQVIA/HyQvia [9, 10]) conducted in healthy participants in the USA, which took place from November 4, 2020 (first participant dosed with study drug) to March 2, 2022 (last participant completed the study). The study was conducted in two parts that ran sequentially (Part 1 followed by Part 2), each with three periods (Fig. 1). The parts differed in the TDL: Part 1 had a TDL of 0.4 g/kg/infusion (equivalent to a volume of 4 mL/kg/infusion), and Part 2 had a TDL of 1.0 g/kg/infusion (equivalent to 10 mL/kg/infusion). In each part, there was an initial screening period of up to 21 days prior to administration of the first dose of study treatment. This was followed by the study treatment period, which lasted until Week 8 or Week 9, as per the treatment arm schedules. The treatment arms were conventional dose ramp-up, accelerated ramp-up, and no ramp-up, which differed according to their dosing schedules. The conventional ramp-up schedule started at 25% of the TDL in Weeks 1 and 2, 50% of the TDL in Week 3, 75% of the TDL in Week 5, and the full TDL in Week 8. This ramp-up schedule was used in the ADVANCE-CIDP 1 study (NCT02549170) [11], and was intended to represent a schedule used to initiate a 4-weekly dosing regimen. The conventional ramp-up used in the current study differs from the schedule recommended in the fSCIG 10% prescribing information in that it includes two initial weekly doses at 25% of the TDL, rather than a single dose. The accelerated ramp-up schedule started at 50% of the TDL in Weeks 1 and 3, with the full TDL administered in Weeks 5 and 9. In the no ramp-up arm, the full TDL was administered at Weeks 1, 5, and 9. Across all treatment arms, each dose was administered on the first day of the week indicated. Upon completion of treatment in Weeks 8 or 9 of Part 1, a safety review team examined tolerability and safety data before commencement of Part 2. The follow-up period lasted up to 16 ± 1 weeks after the last infusion (Fig. 1).
Fig. 1
Study schematic. The conventional dose ramp-up schedule is 1 week longer than that recommended in the fSCIG 10% prescribing information with the inclusion of an additional dose in Week 1. This was done to match the schedule used in the ADVANCE-CIDP 1 study (NCT02549170) [11]. aTime points at which follow-up and end of study occurred are shown by check marks in the Week 24 and Week 25 columns. Recombinant human hyaluronidase was administered prior to the immunoglobulin infusion at a dose of 80 U/g immunoglobulin or 0.5 mL/10 mL immunoglobulin. fSCIG, facilitated subcutaneous immunoglobulin; TDL, target dose level
fSCIG 10% infusions were administered at one or two infusion sites. If two sites were used, they were on opposite sides of the body. Maximum fSCIG 10% infusion volume (excluding rHuPH20 volume) was up to 600 mL/day at one site or up to 1200 mL/day at two sites. The rHuPH20 and Ig components were infused sequentially, starting with rHuPH20.
Study Participants and Treatment AllocationParticipants were required to be: aged 19–50 years at the date of informed consent; male, or non-pregnant, non-breastfeeding females who agreed to comply with contraceptive requirements, or females of non-childbearing potential; determined to be healthy by the study investigator following a physical examination and assessment of medical records; and have a body mass index (BMI) of 18–30 kg/m2. A full list of eligibility criteria is provided in Supplementary Methods. The planned sample size for this study was 48 participants (eight in each of the six treatment arms, with a minimum of three participants in each BMI subgroup [i.e., 18 to < 25 kg/m2 and ≥ 25 to ≤ 30 kg/m2 in each treatment arm]). Each participant was assigned to only one treatment arm. Participant assignment was planned to follow a ratio of 1:1:1.
EndpointsThe primary endpoint was the tolerability of fSCIG 10%. This was assessed as the proportion of participants who completed all initiated infusions without interruption, stoppage, or infusion rate reduction due to a treatment-emergent adverse event (TEAE) that began during the infusion, related to either the rHuPH20 or Ig components. One of the secondary endpoints was the safety of fSCIG 10% infusions. Throughout the study, participants were monitored with clinical laboratory measurements, physical examinations, vital sign measurements, and electrocardiograms. Safety was evaluated by the number of TEAEs and rates of TEAE per participant, per infusion, and per person-year. TEAEs were recorded according to whether they were: fSCIG-related or non-related; non-serious or serious; mild, moderate, or severe; local or systemic; infusion-associated (i.e., any TEAE that began during the infusion or within 24 h following infusion); or led to premature discontinuation from the study. Adverse reactions (ARs) and ARs of special interest were also recorded. The number of participants who developed any binding or neutralizing anti-rHuPH20 antibodies was recorded, and the proportion of participants with anti-rHuPH20 binding antibody titers ≥ 1:160 was summarized between treatment arms [14]. For the determination of binding anti-rHuPH20 antibodies, plasma samples were assayed using a bridging format electrochemiluminescence immunoassay method validated according to current guidance [15] and industry standards [16] at Eurofins Pharma Bioanalytics Services, St Charles, MO, USA. In the event that binding anti-rHuPH20 antibody titers ≥ 1:160 were detected, plasma samples were assessed for neutralizing anti-rHuPH20 antibodies using a validated hyaluronidase enzymatic assay (BioAgilytix, San Diego, CA, USA).
Statistical AnalysisThis study was not designed for statistical hypothesis testing; therefore, the sample size was not based on statistical considerations. All participants who received at least one dose of fSCIG 10% were included in the analysis. Tolerability and safety outcomes were compared between the conventional, accelerated, and no ramp-up arms for TDLs of 0.4 g/kg/infusion and 1.0 g/kg/infusion (i.e., study Part 1 and Part 2) separately.
EthicsThis study was conducted in accordance with the International Council for Harmonisation Guideline for Good Clinical Practice. The study was approved by the IntegReview Institutional Review Board on May 26, 2020.
留言 (0)