
記住我



Seven-week-old male Wistar rats (weighing approximately 180 g) were purchased from the BEIJING HFK BIOSCIENCE CO., LTD, (Beijing, China). Rats were acclimatized for 1 week before the experiment to adapt to the new surroundings. Rats were housed cages and had access to food and water ad libitum. The facility environment had controlled light (12:12 h light/dark cycle) and constant room temperature (23 ~ 25 °C) under conventional laboratory conditions. Rats were assigned randomly into four experimental groups (n = 6–8 for each group): sham sedentary (SHAM), HF sedentary (HF), HF short-term exercise trained (HF-E1) and HF long-term exercise trained (HF-E2). Short-term exercise lasted for 6 weeks (HF-E1) and long-term exercise lasted for 12 weeks (HF-E2) were defined according to the previous study (Guo et al. 2020). All experimental procedures were approved by the Laboratory Animal Welfare Ethics Committee of Military Medical Sciences Academy. All efforts were made to minimize the number and suffering of animals used in these experiments.
Myocardial infarction-induced HF modelExperimental rat MI models were established by permanent ligation of the left anterior descending coronary artery (LAD) of the heart (Li et al. 2021). Briefly, the rats were anesthetized with sodium pentobarbital (30 mg/kg) intraperitoneally (i.p.), endotracheally intubated, and mechanically ventilated with room air (respiratory rate 60–70 breaths/min, tidal volume 2.5 mL). Left thoracotomy between the fourth and fifth ribs was performed to expose and access the rat heart. LAD was identified approximately 2 mm beneath the left atrial appendage and ligated (descending aorta constricted by a 7 − 0 silk suture tied snugly). After ligation, the lungs were re-inflated, after which the chest (6 − 0 silk suture) and skin (4 − 0 silk suture) were closed. Sham-operated rats underwent the same surgical procedure except that their LAD were merely threaded and not ligated. The trachea was extubated as soon as the animals began to recover from anesthesia, and rats were subsequently placed in a warm box (30 ~ 32 °C) for 1 h. Penicillin (80,000 U/kg) was injected intraperitoneally for 3 consecutive days after surgery to prevent infection.
Exercise training protocolRats were trained on a treadmill with individual lanes designed for small animals. One week after the ligation surgery (week 1), rats in the HF-E2 group were subjected to a 4-day adaptive exercise (familiarity period) on a zero-inclination treadmill to minimize potential stress; this consisted of the rats running for 3 min at each of four speeds, 8 m/min, 10 m/min, 12 m/min, and 14 m/min on the first day (a total of 12 min), with the speeds and times of this phase (warm-up) remaining unchanged during the subsequent adaptive and formal exercise training; then the rats ran at 16 m/min for 15 min, followed by a gradual increase of 10 min each day until the fourth day when the rats ran at 16 m/min for 45 min; finally, the rats ran at 8 m/min for 3 min, and the speeds and times of this phase (cool down) remained unchanged during the subsequent adaptive and formal exercise training. The rats in the HF-E1 group started adaptive at week 7 with the same adaptive exercise protocol as the rats in the HF-E2 group, followed by 6 weeks of formal exercise training, whereas the rats in the HF-E2 group underwent 12 weeks of formal exercise training. Formal exercise training for rats in the HF-E1 and HF-E2 groups consisted of running at a speed of 16 m/min for 45 min per day (main exercise) in addition to the 12-min warm-up and 3-min cool down described above, at a frequency of 5 days per week, totalling 1 h per day. Rats assigned to HF group (no exercise) were placed on the static treadmill for a matched stage to minimize the impact of the experimental environment on the results during the entire training (Figs. 1A and 2A).
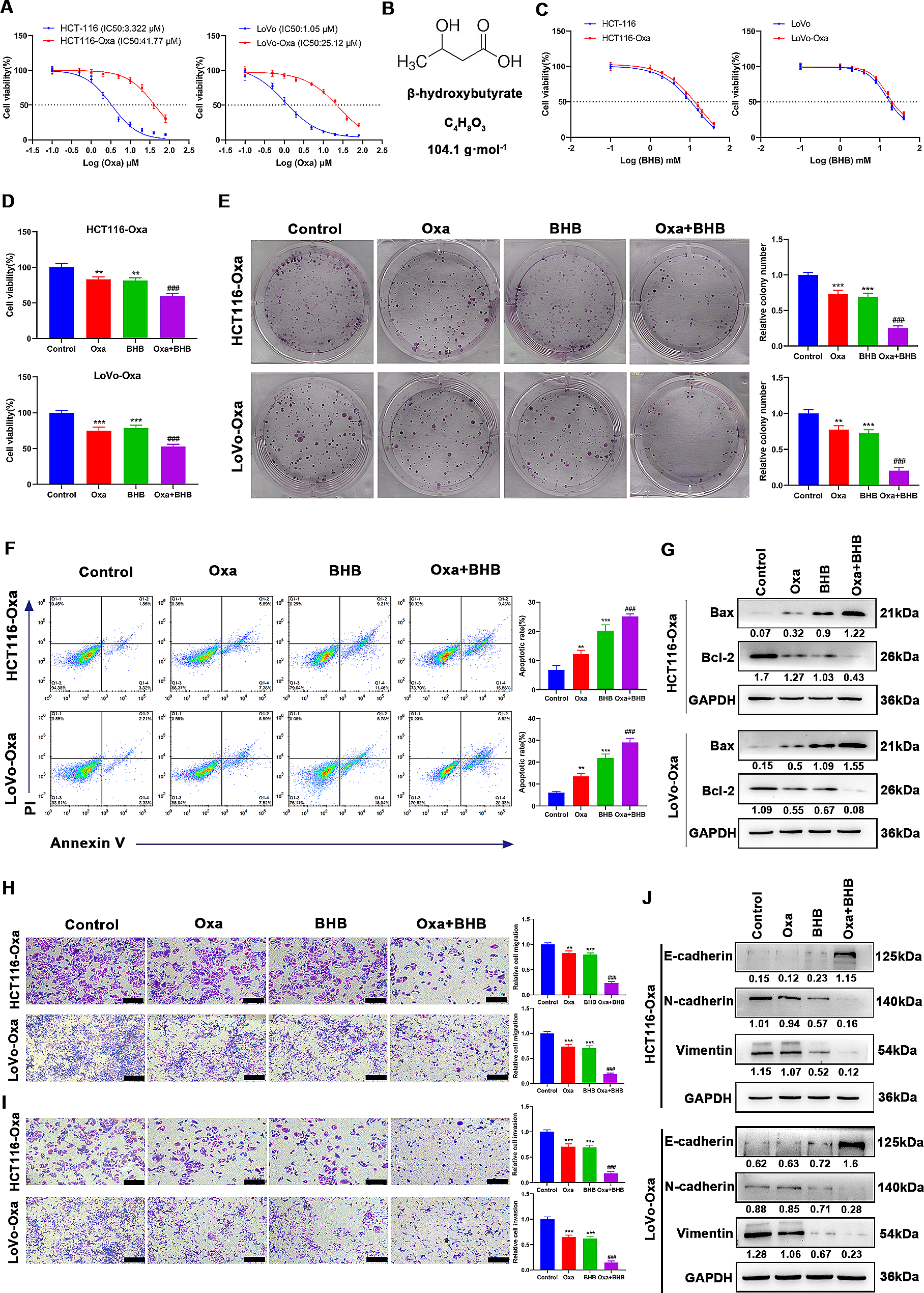
Fig. 1
Pathological remodeling in failing hearts post-myocardial infarction. (A) The timeline illustrates key moments for evaluating left ventricular function in rat groups via echocardiography. (B) Visual comparison of heart morphology across different groups. (C, D) Graphs illustrating changes in heart weight/body weight ratio (HWI) and heart weight/tibia length ratio (HW/TL) among the groups. (E) Echocardiographic images displaying left ventricular function, with wall thickness indicated between yellow arrows. (F, G) Graphical representation of changes in left ventricular ejection fraction (EF) and fractional shortening (FS) before and after LAD surgery Notation ‘*’: denotes statistical comparison between HF and SHAM groups. (H) HE staining images showcasing myocardial tissue in various groups. (I) Masson’s trichrome staining images display myocardial fibrosis in different groups. (J) Quantification of infarct areas based on Masson’s trichrome staining across groups. (K) Electron micrographs representing cardiomyocyte ultrastructure among the groups. (L) Western blot analysis shows the expression of HIF-1α, MCT1, andMPC1 proteins in myocardial tissues, with tubulin as the protein loading control. The student t-test was used for comparisons in the study. Mean ± SD. *: P < 0.05, **: P < 0.01, ***: P < 0.001. n = 6–8
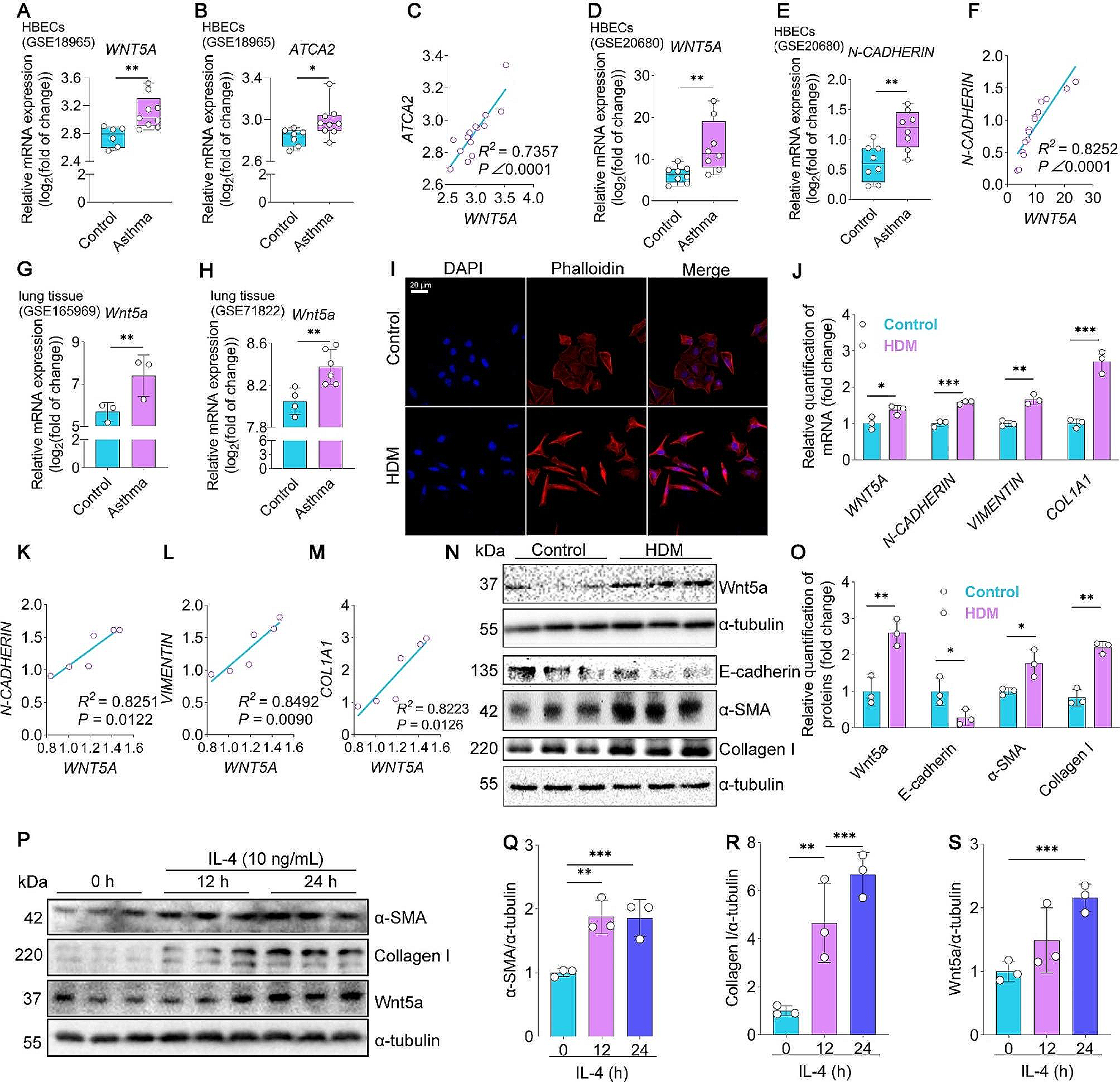
Fig. 2
Treadmill exercise training improves pathologic features of the failing heart. (A) The timeline displays key points for left ventricular function evaluation using echocardiography in each group, conducted before the LAD artery procedure and subsequently during the 2nd, 5th, 10th, and 13th weeks post-LAD procedure. (B) Comparative visualization of heart morphology across groups. (C, D) Graphs depicting changes in heart weight/body weight ratio (HWI)and heart weight/tibia length ratio (HW/TL) among groups. (E) Echocardiographic images illustrating left ventricular function, with wall thickness highlighted between yellow arrows. (F, G) Graphs showing alterations in left ventricular ejection fraction (EF) and fractional shortening (FS) pre and post-LAD. Indicators ‘*’ represent HF vs. HF-E1, ‘&’ symbolize HF-E1 vs. HF-E2, and ‘#’ denotes HF-E2 vs. HF comparisons. (H) HE staining images displaying myocardial tissue variations across different groups. (I) Images of Masson’s trichrome staining of myocardial tissues, indicating differences in fibrosis among groups. (J) Quantification of infarct areas based on Masson’s trichrome staining across groups. (K) Electron micrographs representing the ultrastructure of cardiomyocytes from various groups. (L) Western blot analysis shows the expression of HIF-1α, MCT1, andMPC1 proteins in myocardial tissues, with tubulin as the protein loading control. The one-way ANOVAs followed by Dunnett’s multiple comparison test was used for multi-component comparisons. Mean ± SD. *: P < 0.05, **: P < 0.01, ***: P < 0.001, &: P<0.05, ###: P<0.001. n = 6–8
EchocardiographyNon-invasive cardiac function evaluation was performed by echocardiography in all rats before surgery and at weeks 2,5,10,13 after surgery, respectively (Figs. 1E and 2E). Briefly, an animal anesthesia machine (ventilated 2% isoflurane) was used to anesthetize all rats throughout the surgery. Rats were positioned in the supine position with front paws wide open and ultrasound transmission gel was applied to the precordium. Transthoracic echocardiography was performed using an echocardiographer equipped with a 40-MHz probe, and B-mode images were subsequently obtained in the long and short axis. Cardiac function and structure features such as left ventricular volumes [left ventricular end-diastolic volume (LVEDV) and left ventricular end-systolic volume (LVESV)] and diameters [left ventricular end-diastolic diameter (LVEDD) and left ventricular end-systolic diameter (LVESD)] and fractional shortening (FS) and ejection fraction (EF) were measured and recorded. Left ventricle systolic function was estimated by EF and FS as follows: EF (%) = [(LVEDV − LVESV)/LVEDV] × 100 and FS (%) = [(LVEDD − LVESD)/LVEDD] × 100.
Measurement of tibial lengthsTo calculate heart weight/tibia length (HW/TL), TL in rats anesthetized by a small animal anesthesia machine (1.5% of isoflurane) was measured at the end of exercise training using an animal bone density body composition instrument according to the manufacturer’s protocol.
Tissue preparationAt the end of the training protocol, 48 h after the last exercise session and after 12 h of fasting, all rats were humanely euthanized with ketamine (50 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and body weights (BW) were weighed. All rat hearts were dissected, measured for length and weighed, and myocardial tissues were subsequently collected. Some of the isolated myocardial tissues were fixed with 4% paraformaldehyde/karnovsky fixative solution and others were stored at − 80 °C, these were used for Hematoxylin-eosin (HE) and Masson staining, Electron microscopy analysis and Western blotting, respectively.
Histological staining and analysisFor staining experiments, the formaldehyde-fixed heart tissue samples were washed with water and underwent gradient dehydration and paraffin embedding. Paraffin-embedded heart samples were transversely cut into 5 μm-thick slices onto slides and subsequently stained with HE and Masson’s trichrome. Each sample was observed under the microscope at six randomly selected visions. The Infarct size presented by Masson staining was quantified using Image J software version 1.8 (National Institutes of Health, Bethesda, MD, USA).
Electron microscopy analysisSpecific parts of the heart were taken and fixed in 2% glutaraldehyde and 4% paraformaldehyde in sodium acetate buffer at pH 7.3 for 1 h at room temperature and cut into ~1-mm3 pieces. Samples were washed in phosphate-buffered saline and post-fixed in 2% osmium tetroxide and 1% uranyl acetate for 2 h, rinsed in water, dehydrated in a graded series of ethanol and acetone, and then infiltrated and embedded in Eponate 12 medium. The ultrathin sections were prepared on a Reichert-Jung Ultracut E ultramicrotome (Leica Corporation, Shanghai, China), picked up on copper grids and stained. Images were acquired using a JEM-100CX electron microscope (JEOL Japan Electronics Co., Ltd. Tokyo, Japan) and a 4k digital camera (Gatan Orius 4k X).
Cell culture and hypoxia cell modelH9c2 cells (rat embryonic cardiomyoblast-derived H9c2 cardiomyocytes) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). H9c2 cells were maintained at 37 °C in a 5% CO2 incubator containing complete culture medium (DMEM and 10% fetal bovine serum supplemented with 1% of a penicillin/streptomycin solution) and passaged at approximately 90% confluency. Cell morphologic changes were observed under an inverted fluorescence microscope (CKX53-LP, OLYMPUS CK31MIF-BGU-LED). To establish the hypoxia model, H9c2 cells were cultured in hypoxia incubator with 1% O2, 5% CO2 and 94% N2 for 24 h and 48 h. H9c2 cells in the normoxia group were cultured at 37℃ in a normoxic incubator containing 21% O2 and 5% CO2 and served as the control. Hypoxic H9c2 cells were treated with the HIF-1α activator desferrioxamine mesylate (DFO, 20µM, dissolved in dimethyl sulfoxide, MCE, HY-B0988) (Dziegala et al. 2016) and the HIF-1α inhibitor KC7F2 (20µM, dissolved in dimethyl sulfoxide, MCE, HY-18,777) (Li et al. 2022) for 48 h.
Cell transfectionThree target shRNA plasmids (HIF-1α-RNAi-Easy-shRNA, MCT1-RNAi-Easy-shRNA and MPC1-RNAi-Easy-shRNA) and a negative control (Control-RNAi-shRNA) were developed and synthesized by the manufacturer (Ji Kai Gene Technology Co., Ltd, Shanghai, China), and we subsequently transfected them into hypoxic H9c2 cells using Lipo3000 transfection reagent (GK20006-25, GLPBIO) according to the manufacturer’s instructions. Our transfected plasmid contained the green fluorescent protein gene, and strong fluorescence intensity was observed in hypoxic H9c2 cells under fluorescence microscope, which indicated the high efficiency of cell transfection (Fig. 4A). Briefly, H9c2 cells (5 × 105) were grown in 6-well plates for 24 h. Lipo3000 was diluted using serum-free medium and allowed to stand for 5 min at room temperature. The diluted shRNA was mixed with medium containing Lipo3000 transfection reagent and incubated at room temperature for 15 min, and then the DNA-lipid complex was added into the cells. The transfected cells were grown in hypoxia incubator for 24 h and tested for gene silencing level and expression of each group of indicators.
Cell viability testThe effect of hypoxia on the proliferation of H9c2 cells was detected using the CCK8 Cell Proliferation Kit (Beyotime, Biological Co., Ltd, Beijing, China). H9c2 cells (100 µL) were grown at 2 × 104 cells/mL in 96-well plates overnight. After incubation for 24 h, the cells were treated with hypoxia for 24 and 48 h. After treatment, 100 µL of media enriched with 10% CCK-8 solution was added to the cells through media exchange modes and incubated for 1 h. Absorbance was measured at 450 nm using a Multi-mode Microplate Reader (Molecular Devices, San Jose, CA, USA). Each experiment was performed in sextuplicate, and cell survival rates were expressed as a percentage of the control.
Comparative analysis of ATPThe ATP level analyzed with ATP assay kit (Beyotime, Biological Co., Ltd, Beijing, China). The experimental procedure was consistent with the instruction of the manufacturer. The absorbance readings for the determination of ATP were carried out at 560 nm via the detection of absorbance of a multi-well plate in a Multi-mode Microplate reader (Finalytek, USA). It was able to use this approach to quantify ATP levels in different groups of cells.
Assessment of cell apoptosisAfter hypoxia treatment of H9c2 cells, an Annexin V-FITC/PI apoptosis kit (Keygentec, Nanjing, China) was used for cell staining and flow cytometry following the manufacturer’s instructions. Briefly, cells from each group was washed twice with PBS and 2 × 105 cells were reconstituted with 500µL of binding buffer. Subsequently, 5µL of Annexin V-FITC was immediately added and mixed well. Then another 10µL of PI was added to the cells and incubated at room temperature for 5 minutes in the dark, ready for apoptosis analysis on an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA).
Immunofluorescence analysisCell immunofluorescence was performed by fixing cells with 4% tissue cell fixative (Solarbio, Beijing, China) for 30 min at room temperature, rinsing once with PBS, and then permeabilising with 0.1% Triton X-100 (BioFroxx) for 15 min. Subsequently, the cells were non-specifically blocked with 1% BSA (BioFroxx) for 1 h at room temperature, followed by anti-HIF-1α (20960-1-AP, 1:200, Proteintech), anti-MCT1 (A3013, 1:100, ABclonal) and anti-MPC1 (A20195, 1:100, ABclonal) were incubated overnight at 4 °C. Next, cells were reacted with Alexa Fluor® 594-conjugated goat anti-rabbit IgG secondary antibody (ZF-0516, ZSZSGBBIO) and FITC-labeled goat anti-rabbit IgG secondary antibody (ZF-0311, ZSZSGBBIO) at 1:50 for 30 min at 37 °C protected from light, respectively, followed by reaction with DAPI solution (C0065, Solarbio) for 5 min at room temperature. Finally, imaging was performed immediately on an inverted fluorescence microscope (CKX53-LP).
Western blottingBriefly, protein extracts obtained from isolated myocardial tissue or cultured cardiomyocytes were homogenized in RIPA lysis buffer (Solarbio Life Sciences, R0020, China) containing phenylmethylsulfonyl fluoride (PMSF). The homogenate was centrifuged at 10,000× g for 10 min at 4 °C and supernatant collected, and the total protein concentration was subsequently determined using the BCA Assay Kit (Solarbio Life Sciences, PC0020, China). 5X sample loading buffer was added proportionally to the protein-containing supernatant and boiled (100 °C, 10 min) to obtain protein samples. Protein samples (20 µg) were loaded with standard marker proteins at room temperature and electrophoretically separated (250 mA, 100 min) on polyacrylamide gels (15% Sure-PAGE™, Genscript, China), and then the proteins were transferred onto polyvinylidene fluoride membranes (PVDF, Millipore, USA). Next, membranes were blocked for 2 h at room temperature with 5% defatted milk in TBST (0.1% Tween 20 in TBST) and incubated overnight at 4 °C with the following primary antibodies: HIF-1α (ab179483, 1:1000, Abcam, USA), MCT1 (A3013, 1:1000, ABclonal, China), MPC1 (A20195, 1:1000, ABclonal, China), β-tubulin (380,628, 1:5000, Zen BioScience, China). The membrane was repeatedly rinsed with TBST buffer three times for 10 min each, and incubated with horseradish peroxidase (HRP) conjugated IgG antibody-Goat Anti-Rabbit IgG H&L/HRP (bs-40295G-HRP, 1:10000, Bioss, China) for 1 h at room temperature. After incubation with the secondary antibody, the membrane was repeatedly rinsed three times for 10 min each with TBST buffer and treated with enhanced chemiluminescence reagent (ECL, WBKLS0500, Millipore, USA). Subsequently, protein bands were visualized via chemiluminescent detection in a gel image processing system (Amersham Imager 680, CTL, USA), and quantified by densitometry using Image J analysis software version 1.8. Targeted bands were normalized to the relative expression of cardiac β-tubulin.
Statistical analysisThe results are reported as the Mean ± SD. All data were statistically analyzed and graphed using SPSS software (version 22.0) and GraphPad Prism (version 8.0). The unpaired two-tailed Student t-test was used for comparison between two groups, and one-way ANOVAs followed by Dunnett’s multiple comparison test was used for multi-component comparisons. All experiments were conducted at least three biologically independent replicates. Statistical significance was accepted at *p < 0.05, **p < 0.01, ***p < 0.001.
留言 (0)