Subjects
Synovium specimens were obtained from knee OA patients (4 men and 6 women) who were undergoing joint replacement or synovectomy. All patients studied were Iranian, and the mean age of OA patients was 57.44 ± 11.45 years. Tissue samples were collected from Shariati and Laleh hospitals, Tehran, Iran. The patients were end-stage, and the diagnosis was performed based on the 1986 criteria of the American College of Rheumatology (ACR) [16, 17]. An informed consent form to participate in this research was signed by all subjects. The study protocol was reviewed and approved by the ethics committee of Iran University of Medical Sciences (IR.IUMS.FMD.REC.1398.123) and Tehran University of Medical Sciences (IR.TUMS.VCR.REC.1397.037).
Cell isolation and culture
The knee synovial tissue samples of OA patients were collected from the surgery department in a transporter media containing 15 ml Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Life Technologies, USA) culture medium with 1% penicillin-streptomycin and transferred to the cell culture lab. The tissues were soaked in sterile phosphate-buffered saline (PBS, Gibco Invitrogen, USA) pH (7.3–7.4), alcohol (ethanol 70%), and finally with PBS containing 1% penicillin-streptomycin.The collected tissues were minced into 1 mm X 1 mm pieces in a sterile microplate containing DMEM. The collagenase VIII (1 mg/mL, Sigma–Aldrich, USA) was added to a 50 ml conical centrifuge tube (SPL, Life Sciences, Korea), which contained the dissected synovial tissue fragments, and incubation was performed in a shaker incubator for 80 min 37 °C to promote isolation of FLSs. The samples were vortexed and resuspended two or three times during the incubation.
Then the digested tissues were centrifuged for 10 min at 1000 g, the supernatant was discarded, and the cell pellets were resuspended in 1 mL of complete DMEM before transferring them into two T25 flasks (SPL, Life Sciences, Korea) containing 4 mL of complete DMEM media. The T25 flasks were moved to an incubator with 5% CO2 at 37 °C in a humidified atmosphere. A homogeneous population of FLSs was obtained after three passages. Consequently, for later assessments and interventions, FLS cell passages three through six were employed.
Identification of FLSs by immunofluorescence staining and flow cytometry
To confirm the isolated cells’ fibroblastic origin, immunofluorescence staining was used. Initially, FLS cells were seeded into a 24-well culture plate and incubated for twenty-four hours at 37 °C. After incubation time the cells were washed with PBS. Next, the cultured FLSs were incubated in cold methanol to fix the cells. Subsequently, the cells underwent a PBS wash and were incubated for one hour on a shaker with a blocking buffer (PBS with Triton-X100 contains 1% BSA). Then, incubation of the cells was performed with the primary antibody, i.e., anti-fibroblast surface protein antibody (ab11333, Abcam, UK), overnight at 4 °C. The secondary antibody, i.e., sheep anti-mouse Ig (human Ig absorb)-FITC conjugated (Ibn Sina, ARI2011F, Iran), was then used to incubate for 60 min at room temperature in a dark environment. To counterstain the nuclei, the 4′, 6-diamidino-2-phenylindole (DAPI) was implemented. Lastly, an inverted fluorescence microscope was implemented for the morphological assessment of stained FLSs.
Several of the primary cell surface CD markers, such as CD13, CD44, CD68, and CD90 [18, 19] were investigated by the flow cytometry technique to further validate the fibroblasts as FLSs. To achieve this goal, FLSs were gathered and subjected to three PBS washings. After that, incubation of the FLSs was performed with the fluorescein isothiocyanate (FITC)-conjugated antibodies against the mentioned surface CD markers at 37 °C for one hour. The antibodies used included anti-CD13 antibody (ab227663), anti-CD44 antibody (ab6124), anti-CD68 antibody (ab31630), and anti-CD90 antibody (ab225), and all of them were purchased from Abcam Inc. (Cambridge, UK). Other FACS analyses used unlabeled cells as the negative control.
FLS cell grouping and intervention
Every FLS sample was separated into the following 3 study groups: (i) the Untreated group (no treatment), (ii) the LPS group (100 ng/mL), and (iii) the treatment group (TAK-242 (32 µM) + LPS (100 ng/mL)). For determining the effects of TAK-242 on FLSs, pre-treatment of OA-FLSs with TAK-242 (32 µM) was performed in culture media for one hour, and then, the cells were stimulated by LPS (100 ng/mL) for six hours.
RNA extraction and cDNA synthesis
An RNA extraction kit (SinaColon Co. Tehran, Iran) was used to extract total RNA from each of the three groups according to the manufacturer’s manuals. Then, the purity and yield of extracted RNA were investigated via a NanoDrop spectrophotometer (NanoDrop ND-2000 C Spectrophotometer, Thermo Fisher Scientific, USA) at 260/280 nm absorbance. The extracted RNAs were kept at −80 °C for the next molecular assessment. The first-strand complementary DNA (cDNA) synthesis process was applied to the total RNA using an RNA reverse transcription kit (RT-ROSET, ROJE, Iran).
Quantitative real-time PCR
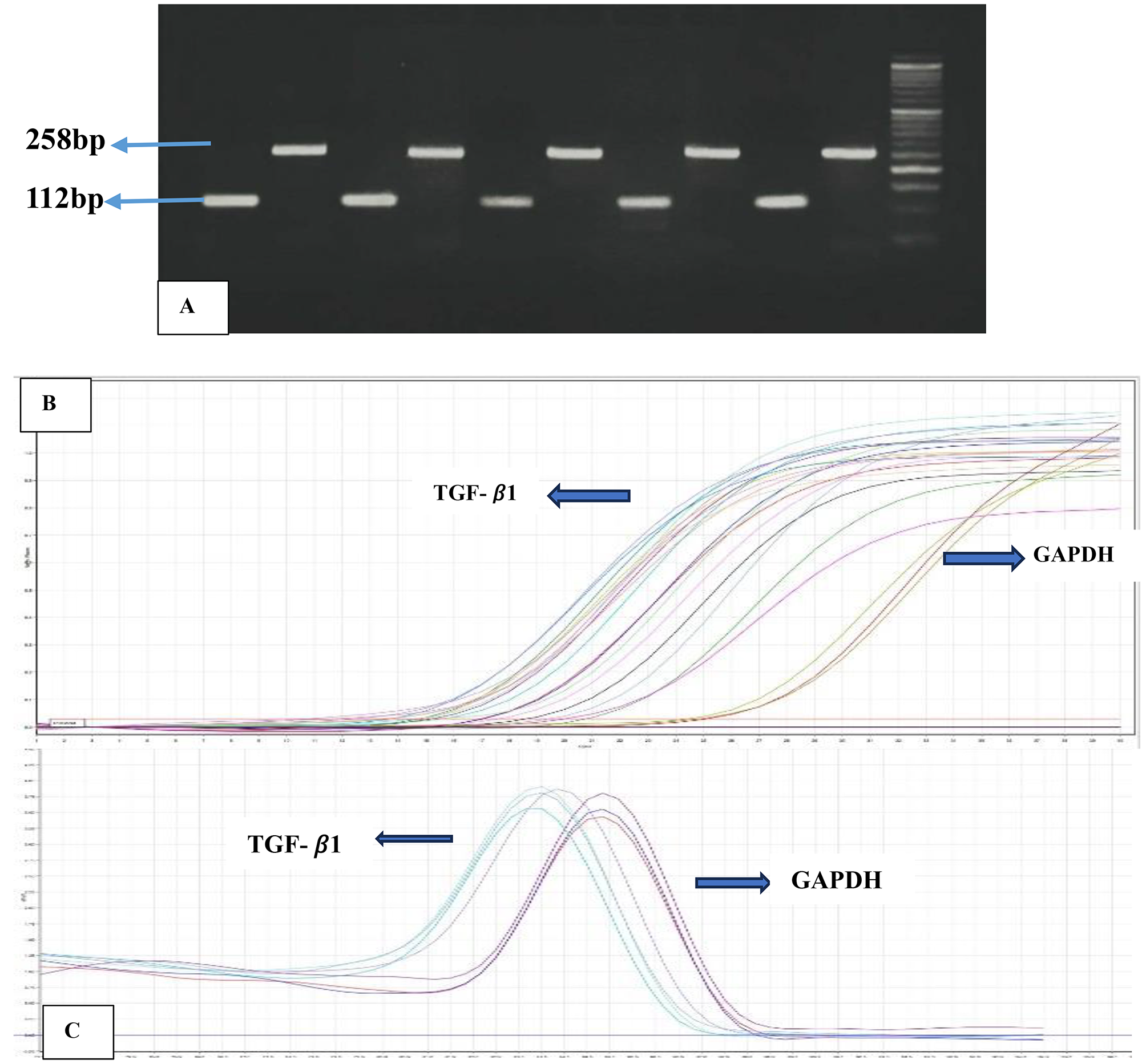
The quantitative polymerase chain reaction (qPCR) was adopted using the SYBR Green gene expression master mix and Applied Biosystems StepOnePlus Real-Time PCR System (Foster City, CA, USA). The comparative Ct (2-ΔΔCt) method was used to calculate the relative mRNA expression levels of the target genes to Glyceraldehyde-3- phosphate dehydrogenase (GAPDH). The primers and the related sequences are provided in Table 1.
Protein extraction and western blot
The IκBα and pIκBα protein levels were measured using Western blot analysis. Following a six-hour treatment of the cells with TAK-242 and LPS, the samples were rinsed with ice-cold PBS before being subjected to total protein extraction using the radioimmunoprecipitation assay (RIPA) lysis buffer. This buffer contained whole EDTA-free protease inhibitor cocktail tablets (Roche, Germany). To determine the protein concentrations, the Lowry method was employed. Following that, they were separated using an equivalent volume of protein extracts (50 µg) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 90 V for 2.5 h. Subsequently, the separated proteins were transferred to a PVDF membrane (Thermo Scientific, USA) at 100 V for 90 min. The membrane was incubated with 5% skim milk (Sigma-Aldrich, USA) in 1 X Tris-buffered saline with Tween (TBST) for 60 min at room temperature, to block the free spaces. After that, primary antibodies (anti-IκBα (ab97783, 1:1000), anti-pIκBα (2859, 1:1000), and anti-β-actin (ab8226, 1:1000), Abcam, UK), were used to incubate the membrane overnight at 4 °C. Following the incubation, the membrane was washed with TBST, to eliminate the free antibodies. Horseradish peroxidase-conjugated secondary antibody (PZ5610, 1:3000) was then added, and the blots were incubated for two hours at room temperature. Following the washing of the blots, the protein bands were seen using an enhanced chemiluminescence detection reagent (ECL, GE Healthcare, USA). β-actin was used as an internal control to normalize the results, and the data were semi-quantified using Image J software (NIH, USA).
Statistical analysis
SPSS software version V26.0 was utilized for analyzing the findings. Also, the GraphPad Prism software V8.0 was used for designing the graphs. Initially, the normality of the data was investigated by the normality test, and it was observed that the data were not normally distributed (Kolmogorov-Smirnov < 0.05). To compare several paired groups, the Friedman test was employed. In order to compare two paired groups, the Wilcoxon signed-rank test was employed. The quantitative data are presented as mean ± standard error of the mean (SEM), and P-values less than 0.05 were set to be statistically significant.







留言 (0)