
記住我
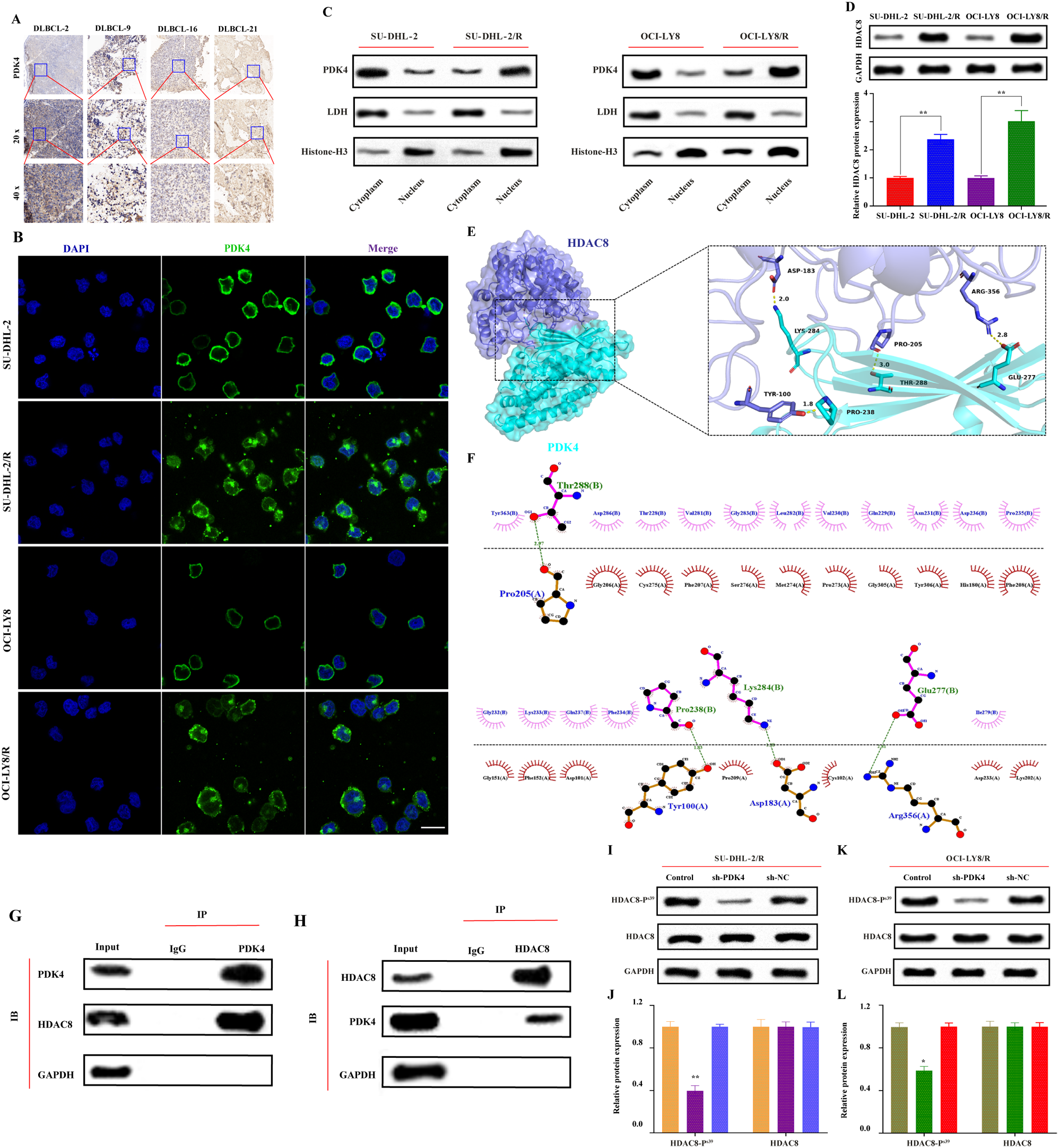
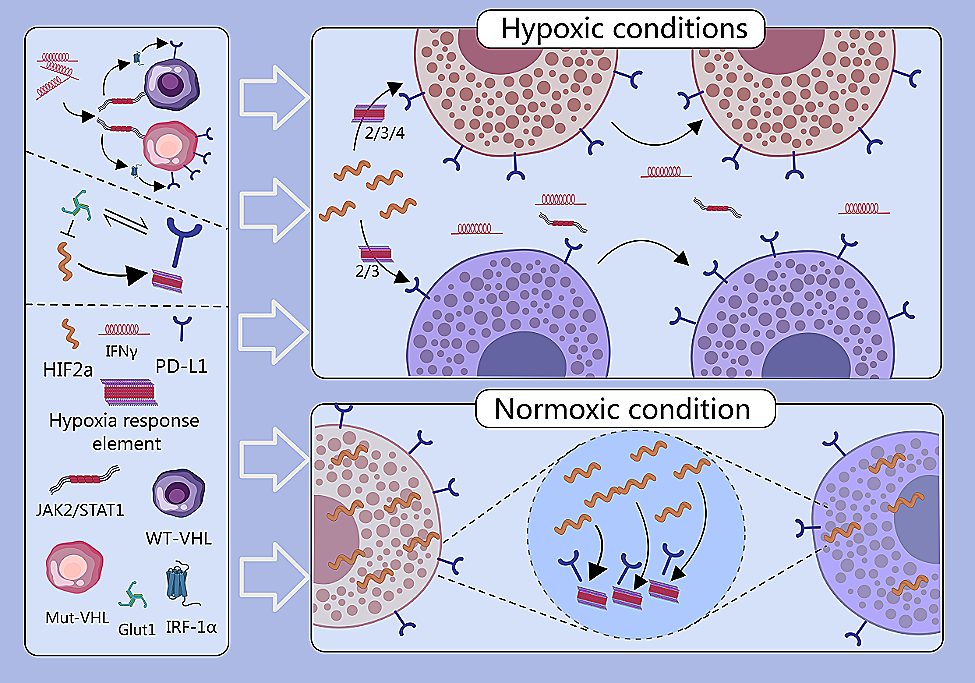


Given the presence of somatic mutations relevant to cancer in a subset of patients with lung adenocarcinoma (LUAD), we often encounter significant challenges when attempting to apply targeted therapies. To broaden the scope of precision medicine to include patients with clinically actionable genetic alterations (CAGAs), we used a comprehensive strategy to classify patients with LUAD. Our initial classification scheme emphasized the identification of primary mutations in essential oncogenes, such as EGFR, KRAS, BRAF, ERBB2, and MET (exon skipping), as well as in oncogenic fusion genes, including ALK, ROS1, NRG1, RET, NTRK, and FGFR. Identifying these mutations is particularly important for cancer therapy because targeted treatments specifically designed for these mutations have demonstrated significant therapeutic benefits [1]. Therefore, we selected LUAD cases from 938 patients using WES and poly(A) RNA-seq dataset. From the subset that did not possess these CAGAs (n = 420, termed non-CAGAs), we selected 174 cases for WGS and H3K27Ac ChIP-seq analyses (Fig. S1A). Of note, driver mutations in genes including EGFR, KRAS, BRAF, and ERBB2 were identified in 476 cases, representing 50.7% of the entire cohort (Fig. S1B). Importantly, a higher frequency of mutations was observed in the non-CAGA cohort (Fig. S2A, and the oncoprints shown in Fig. S2B and C), suggesting that these variants may not serve as specific markers for non-CAGA cases but rather indicate a general elevation in mutation frequency. Furthermore, the CNV and SV landscapes revealed hotspots associated with the CDK4/MDM2 loci, where copy number amplification was observed. This may be partly explained by complex chromothripsis events characterized by extensive copy number amplification (Fig. S3).
To elucidate the distinct characteristics between normal and tumor tissues, we conducted H3K27Ac ChIP-seq analysis of seven non-CAGA LUADs. Adjacent matched tissues were used as normal controls for comparison. The PCA results indicated that, while the adjacent tissues manifested homogenously, the lung adenocarcinoma samples exhibited diverse features (Fig. S4). Subsequently, we performed WGS and H3K27Ac ChIP-seq in and 174 patients without CAGAs (non-CAGAs, see Fig. S2B and C and Table S1) and 45 patients with CAGAs to comprehensively investigate the potential roles of super-enhancers in our LUAD cohort (QC data are summarized in Fig. S5 and Dataset S1).
To explore the direct correlation between the formation of super-enhancers and genomic structural variants, and to better understand their molecular interplay in disease mechanisms, we employed Manta analysis to identify genomic breakpoints from WGS and ROSE analysis to identify super-enhancer regions from ChIP-seq and obtained the genomic loci where these two sets of data overlapped (Fig. 1A, super-enhancer and structural variant regions summarized in Dataset S2-3, 4–5, respectively). Although the total number of loci in the entire dataset was 67,349 and 69,991, we found that only a small fraction, 700 (~ 1%), showed overlapping regions (Fig. S6, genome coordinates listed in Dataset S6-7), suggesting that structural variants play a confined role in specific regions as direct triggers for the formation of super-enhancers in non-CAGAs LUAD. A noteworthy finding was that when focusing on regions where super-enhancers and structural variants overlapped, the frequency of overlaps per patient in non-CAGA was substantially higher than that in CAGAs LUAD (Fig. 1B). This suggest that in some instances, the concurrent presence of super-enhancers and structural polymorphisms may act as discriminating factors for non-CAGA LUAD. Furthermore, all pathways were significantly associated with cancer-related processes in the non-CAGA LUAD group (Fig. 1C). Conversely, cancer-related pathways were not consistently observed for gene groups located near the super-enhancers and structural variant regions alone (Fig. S7). Finally, we confirmed the formation of super-enhancers accompanied by structural variants in genes such as BAX, CCND1, CDK4, EGFR, ERBB2, FOXO3, RXRA, and STAT3, which are all frequently related to NSCLC (Fig. 1D, Fig. S8).
Fig. 1
Intersection of SVs with SEs in non-CAGAs LUAD. A Bioinformatics methodology employed to detect SEs that are potentially regulated by SV events. Given the extensive perturbations to the 3D genomic architecture associated with SV events, a significant association is inferred when a 20-kb region surrounding the genomic breakpoint exhibits a substantial overlap with an SE region. A comprehensive description of the methods can be found in the method section. B Comparison of log2 frequency of SV events per sample (left side) and SE-SV overlaps per sample (right side) between CAGAs LUAD (n = 45) and non-CAGAs LUAD (n = 174). Statistical analysis was conducted using a two-sided t-test. * P < 0.05, ns: not significant. C KEGG pathway enrichment analysis on gene clusters annotated as SE regions with concurrent SVs in non-CAGAs LUAD samples. The statistical significance of the enriched pathways was determined using the enrichKEGG function from the clusterProfiler R package. The background gene set was defined as genes annotated with the SV regions alone. Comparable results were observed when using genes annotated with SE regions alone as the background (data not shown). False discovery rate (FDR) was calculated using the Bonferroni correction method, and the q-scores (qscore) were represented as -log10(FDR). The 20 enriched pathways are displayed (FDR < 0.05). D The gene clusters obtained from the top 5 enriched pathways in non-CAGAs LUAD. The counts of individual genes annotated in regions where SE and SV overlaps were provided
To explore the potential impact of structural variants on gene expression in our dataset, we conducted an integrated analysis of RNA sequencing data and structural variants. Most genes showed no significant changes in expression levels (black dotted line in Fig. S9A). However, a subset of genes (n = 632) exhibited elevated expression levels, which may be associated with the presence of structural variants (red dotted line). Conversely, 170 genes exhibited decreased expression levels (blue dotted line). Notably, no significant differences in outliner gene expression (red dots, n = 20) were observed between the non-CAGA and CAGA cohorts, suggesting that structural variants alone cannot be used to distinguish between non-CAGA and CAGA cases (Fig. S9B). Hence, identifying the genomic regions where both super-enhancers and structural variant coexist might offer insights into the cancer-related attributes of non-CAGAs LUAD. These findings suggest that the genes identified in these regions have potential therapeutic implications.
Impact of gene expression on super-enhancer formation accompanied by structural variants in non-CAGAs LUADTo investigate distinct cellular or tissue signatures within LUAD through transcriptional profiling, we performed a clustering analysis on the entire RNA-seq dataset comprising 938 cases. This analysis revealed that a specific subset of non-CAGA LUAD cases exhibited prominent characteristics similar to those of limbal and corneal epithelial stem cells. In contrast, the EGFR mutation-positive group was markedly enriched in Type II pneumocytes and epithelial progenitor cells as shown in group 1 and 3, respectively (Fig. S10 and Table S2). To decipher the super-enhancer and structural variant landscape in our non-CAGAs LUAD cohort and better understand its impact on gene expression, we performed a peak-to-gene links analysis [29], by correlating H3K27Ac peaks within 0.5 M bp of the gene promoter with the expression of the gene (n = 142). In this analysis, 10,683 genes were identified to have a significant quantitative correlation with H3K27Ac peaks (FDR < 0.05, top 1,000 lists summarized in Dataset S8). A notable observation from our data suggests a positive correlation among gene clusters annotated as super-enhancer regions that also have accompanying structural variants (Fig. S11). Strikingly, genes such as ERBB2 and EGFR, which are recognized as representative driver genes in LUAD, ranked prominently in this assessment (Fig. 2A, B, Table 1). Moreover, although CDK4 and MDM2 have been demonstrated to be involved in lung cancer, their roles as therapeutic targets have not yet been firmly established [30]. Regardless, they were ranked the most prominent in this assessment (Fig. 2C, D, Table 1). In a limited number of non-CAGA LUAD cases involving the ERBB2, EGFR, CDK4, and MDM2 genes, we identified events where gene expression was induced to a considerable extent that they were deemed outliers (Fig. 2A-D). We confirmed that the super-enhancer and structural variants overlaps, which served as the origin of the genomic rearrangements, were present in all these cases (Fig. 2E-G). Importantly, structural variations associated with super-enhancers did not exhibit extensive copy number amplification, albeit with a moderate gain in copy number (Fig. 2E-G). Finally, to elucidate the differences in gene expression patterns and pathway engagements, particularly between those with super-enhancers and structural variants in genes including ERBB2, EGFR, KRAS, CCND1, MDM2, and those primarily displaying copy number alterations (CNAs), we conducted a comparative expression analysis. This analysis distinctly identified the chemokine activity pathway as significantly involved in cases with super-enhancers and structural variants, as highlighted in Group 4 (Fig. S12, Table S3). These findings indicate that H3K27Ac peaks provide a more explicit marker for gene expression amplification associated with the formation of super-enhancers concomitant with structural variants. Therefore, our analysis of the super-enhancer and structural variant landscape successfully identified gene clusters with strong correlations to expression levels. However, in instances where super-enhancer and structural variant overlaps were present, we observed an exceptionally aberrant elevation in gene expression.
Fig. 2
Gene expression on SE formation accompanied by SVs in non-CAGAs LUAD. A-D Correlation of H3K27Ac peaks with the expression of the genes (n = 142). The genes annotated as both SE regions and SVs were extracted (SE-to-gene links analysis). The straight line (blue) is to represent the best fit for the data points based on the least squares method. A linear model was used for the smoothing. To measure outliers, the local outlier factor (LOF) method was employed. For each data point, the 10 nearest data points were identified. A comprehensive description of the methods can be found in the supplementary methods. Each data point is represented in a heatmap according to LOF scores. E–G Circos plots of individual non-CAGAs LUAD samples. Representative cases detected by the LOF method are shown. SE regions are indicated by blue bands. The chromosomal number of the origin region, where the SE and SV overlap, is denoted in red. DEL: deletion, DUP: duplication, INV: inversion, TRA: translocation, INS: insertion, SE: super-enhancer. The absolute CNV calls were indicated in an outer ring of the Circos plots
Candidates of driver mutations driven by exceptionally aberrant elevation in gene expressionOur SE-to-gene link analysis revealed a group of genes displaying remarkably aberrant expression that are compelling candidates for driver mutations driven by both super-enhancers and structural variants. Therefore, additional driver genes may need to be identified. Indeed, genes such as FRS2 and CAV2 may emerge as candidates (Fig. S13, the peak-to-gene link analysis for all other candidate genes, as shown in Fig. S14 and the Circos plots shown in Fig. S15). FRS2 (Fibroblast Growth Factor Receptor Substrate 2) plays a critical role in activating the MAPK and PI3K signaling pathways, which are essential for cell proliferation, migration, and survival [31]. It has been identified as oncogenic and is amplified in high-grade serous ovarian cancer, highlighting its potential as a driver gene in oncogenesis [32]. Similarly, CAV2 (Caveolin 2) is implicated in cancer progression; genetic variants leading to high CAV2 expression have been shown to promote pancreatic cancer progression and are associated with poor prognosis [33]. Furthermore, CAV2 influences focal adhesion and extracellular matrix organization pathways, underscoring its role in tumor development and metastasis [33]. In summary, this analysis suggests that FRS2 and CAV2 are involved in the molecular dynamics of non-CAGA LUAD. A deeper understanding of their molecular mechanisms may provide insights into potential therapeutic strategies.
Chromosomal structure of super-enhancer and structural variant overlapped ERBB2 gene locusConsidering the notably aberrant increase in gene expression driven by super-enhancer formation associated with SV events, it is noteworthy that such super-enhancer and structural variant overlapping cases were observed in 40.8% of patients with non-CAGA LUAD (Fig. 3A, gene clusters on KEGG pathway enrichment analysis: FDR < 0.05). Although a small patient group with super-enhancer and structural variant formation was observed for the ZFP36L1, DDIT4, and MIR21, unique super-enhancer and structural variant formations were observed in individual patients (Table S4). Among these, we focused on non-CAGAs LUAD cases displaying super-enhancer formation around ERBB2, comprising 1.15% of non-CAGA LUAD patients (Table S4). To further evaluate the validity of ERBB2 as a potential drug target in non-CAGA LUAD cases, we conducted H3K27Ac ChIP-seq analysis in HER2-overexpressing LUAD cases verified by IHC and RNA-seq; however, its relationship with genomic amplification remains unclear [34]. These analyses were performed using patient-derived xenograft (PDX) models established at the NCC Japan [34, 35]. Extensive super-enhancer formation in the HER2 region was indeed observed (Fig. 3B, Fig. S16A). This super-enhancer formation led to marked overexpression of HER2, as evidenced by both transcripts (Fig. S16B), and protein levels (Fig. S16C). To investigate the activation mechanisms of overexpressed ERBB2, we analyzed the same PDX samples as previously mentioned: one harboring an EGFR activating mutation L858R, sample #1, and the other exhibiting ERBB2 overexpression, sample #2. This analysis was performed utilizing both mass spectrometry and reverse-phase protein array methodologies. Although we confirmed ERBB2 overexpression (Fig. S17A), we did not observe a significant increase in phosphorylated ERBB2 at Y1248—a well-established marker of ERBB2 activation (Fig. S17B). However, we found that phosphorylation levels of ERK1/2 and the S6 ribosomal protein within the PI3K-AKT-mTOR pathway were found to be comparable in both PDX samples (Fig. S17C). Despite the limited number of samples, this suggests that there are common activation mechanisms in ERBB2 overexpressed cases that do not depend on its Tyr-1248 phosphorylation, indicating alternative pathways could be involved in ERBB2-driven signaling [36]. Importantly, drug testing using the pan-HER inhibitor, poziotinib exhibited a significantly promising effect, whereas afatinib showed no antitumor effect. In contrast, trastuzumab deruxtecan (T-DXd) induces significant tumor shrinkage in a dose-dependent manner [34]. These findings suggest that ERBB2-targeting therapies, particularly poziotinib and T-DXd, could be effective therapeutic options for LUAD with super-enhancer formation around ERBB2.
Fig. 3
Extensive chromosomal rearrangement coincident with SE formation in the ERBB2 locus. A Pie chart illustrating the proportion of non-CAGAs LUAD patients (n = 174) with SE formation associated with SV events. The frequency of the relevant cases displayed as numbers around the outer edge of the pie chart are shown. We counted the number of cases that overlapped with genes extracted through KEGG pathway enrichment analysis (FDR < 0.05). B Genome browser view of H3K27Ac ChIP-seq tracks for PDX model in LUAD. The region on chromosome 17: 39,650,000 to 39,800,000, with a center on the ERBB2 gene is shown. C Integrative visualization of Hi-C, WGS, and ChIP-seq data in a sample where ERBB2 expression was identified as an outlier. The figure presents a broad region on chromosome 17, spanning from 32,500,000 to 42,500,000. In this Hi-C analysis with a triangular view, chromosomal rearrangements are mainly represented as heatmaps, which provide a visual representation of the frequency of interactions between separate genomic regions within the cell nucleus. A missing region in the heatmap represents areas that have not been annotated in the reference genome. In the WGS track, connected breakpoints detected by Manta are linked with light blue lines. The genome browser view of H3K27Ac ChIP-seq tracks was shown at the bottom. A region where high contact frequency (Hi-C), clustering of genomic breakpoints (WGS), and peaks in H3K27Ac signal (ChIP-seq) were observed in distal genomic regions was marked with a light green bar at the bottom. D The assembly graph as a single node style demonstrates the de novo assembly of chromosomal rearrangement around the HNF1β-ERBB2 loci. Two distinct paths show the connection between the HNF1β and ERBB2 genes. The HNF1β gene is represented in green, while the ERBB2 gene is displayed in blue. A 25 kb genome size bar is shown for reference
To delve deeper into large-scale chromosomal structural changes and interactions, we conducted Hi-C analysis of cases exhibiting extensive super-enhancer formation surrounding the ERBB2 gene. Genomic alterations coinciding with H3K27Ac peaks were corroborated by the Hi-C results, as demonstrated by altered genomic organization (Fig. 3C). To directly identify the bona fide structural variants, we conducted de novo assembly using long-read sequencing with the PacBio Sequel II platform in conjunction with Hi-C data obtained from the same specimen (Fig. S18A, referring to Materials and methods). Upon analysis, we observed that the ERBB2 gene loci were situated in closer proximity (~ 125 kb) to the HNF1β gene loci compared to their respective positions in the standard GRCh38 reference genome (~ 1.9 Mb apart) (Fig. 3D) while preserving contiguity (Fig. S18B). This observation suggested that a structural variant event was responsible for the rearrangement of the ERBB2-HNF1β gene loci (Fig. 3D). This comprehensive analysis not only elucidates the complex genomic landscape of non-CAGA LUAD, but also highlights the potential of ERBB2-targeting therapies for a subset of patients with specific super-enhancer formations.
Targeted chromosomal rearrangements between ERBB2 and HNF1β loci in cultured cellsTo directly determine whether the characteristic structural abnormalities obtained from the aforementioned results led to aberrant gene expression, we induced genomic structural abnormalities in cultured cells using the CRISPR-Cas9 system. WGS and Hi-C analyses revealed highly complex structural abnormalities in the ERBB2 region. Meanwhile, from the Hi-C analysis results (Fig. 4A), we identified common chromosomal inversions in the ERBB2 and HNF1β gene loci, respectively (Fig. 4B-D). Therefore, we designed gRNAs targeting the regions adjacent to these two breakpoints and attempted to induce chromosomal inversions in HBEC3-KT and HSAEC1-KT cells (Fig. S19). These cell lines, immortalized with CDK4 and hTERT, represent human bronchial and small airway epithelial cells, respectively, and neither form colonies on soft agar nor initiate tumor growth in mice [37, 38]. No oncogenic mutations in EGFR have been detected in HBEC3-KT using WES [39]. To confirm specific inversions, we employed T7EI assays and sequencing techniques (Fig. S20-21). Chromosomal inversions require simultaneous double-strand breaks at two distinct locations. When double-strand breaks were simultaneously induced, approximately 0.20–0.69% of the cells displayed an increase in HER2 expression, as confirmed by FACS (Fig. 5A-B, Fig. S22A-B) and RT-PCR (Fig. S23). This is comparable to the reported frequency of chromosomal inversions of approximately 1–8% [40, 41]. This increase was also observed with gRNAs targeting different sequences, albeit in the proximate regions (Fig. 5C-D, Fig. S22C-D). Conversely, upon inducing a break at only one site, we observed no significant difference in HER2 expression compared to baseline, with an approximate frequency of 0.01–0.04% (Fig. 5E-J, Fig. S22E-J, summarized in Fig. 5K, Fig. S22K). These results indicate that an increase in HER2 expression occurs only when double-strand breaks are induced in both ERBB2 and HNF1β genomic regions, strongly suggesting that the observed genomic structural abnormalities directly impact HER2 expression.
Fig. 4
Integrated visualization of Hi-C and ChIP-seq data. A Table summary of the case used in the integrated analysis of Hi-C and ChIP-seq. B-D The displayed region on chromosome 17, which covers positions 37,034,000 to 40,422,000, lies between the HNF1β and ERBB2 gene loci. The region enclosed by the green dashed line indicates the proximity of the HNF1β and ERBB2 gene loci. The case shown in panel D was used as a negative control in which ERBB2 is not overexpressed. dJapanese PDX [35]. eVerified by IHC and RNA-seq [34]
Fig. 5
Genomic inversion induced by CRISPR-Cas9 mediated double-stranded break at HNF1β and ERBB2 gene loci. Cas9-inducible HBEC3-KT cells were transfected with the combinations of guide RNAs, A gRNAs #1 and #2, B gRNAs #2 and #3, C gRNAs #1 and #4, D gRNAs #2 and #4, E gRNA #1, F gRNA #2, G gRNA #3, H gRNA #4, I Control gRNA, J Buffer. Cells within the red-framed region were designated as HER2 overexpressed cells. K In each experimental condition, the proportion of HER2 overexpressed cells (%) within the red-framed region was summarized
Significance of outlier genes in clinical outcomesOur SE-to-gene link analysis, prioritized by the top six genes (CDK4, ERBB2, MDM2, FRS2, EGFR, CAV2), identified a set of genes displaying markedly aberrant expression patterns, indicative of potential driver mutations (Table 1). To evaluate the clinical implications of gene overexpression in the absence of somatic mutations, we analyzed its correlation with recurrence-related clinical outcomes. In patients with non-CAGA LUAD (n = 312), the presence of pronounced aberrant gene expression elevation, as ascertained by gene expression outlier analysis (refer to Materials and methods), was associated with significantly decreased RFS compared to those without such elevation (Fig. 6A). Moreover, these results were observed irrespective of driver mutations in the LUAD cohort (n = 1,147, Fig. 6B). Additionally, comparable results were obtained when the entire set of 26 genes extracted from the SE-to-gene link analysis (Table 1) was considered as the target group (Fig. 6C-D). Among the 26 genes, all LUAD cases with outliers in the 23 gene groups exhibited an increased risk of recurrence, particularly with FGF3, FGF4 and FGF19, which are involved in recurrence risk (Fig. 6E). These findings underscore the robustness of the gene set derived from the super-enhancer and structural variant landscape analyses and imply that regardless of the presence or absence of driver gene mutations, such as CAGAs, the identified genes possess clinical significance as prognostic factors for predicting postoperative outcomes in LUAD.
Fig. 6
Aberrant gene expression is associated with an increased risk of postoperative recurrence in LUAD. Kaplan–Meier analysis was performed for LUAD patients with identified outlier genes, focusing on RFS in cases exhibiting abnormal gene expression as outliers (refer to Materials and methods). A non-CAGAs LUAD cases stratified by the top 6 outlier genes. B all LUAD cases stratified by the top 6 outlier genes. C non-CAGAs LUAD cases stratified by all 26 outlier genes. D all LUAD cases stratified by all 26 outlier genes. Red lines: LUAD cases with aberrant outlier gene expression. Blue lines: LUAD cases without aberrant outlier gene expression. E A radar chart illustrating the prevalence of the 23 genes in cases containing at least one outlier gene. Red line: non-CAGAs LUAD cases; Orange line: all LUAD cases
留言 (0)