
記住我
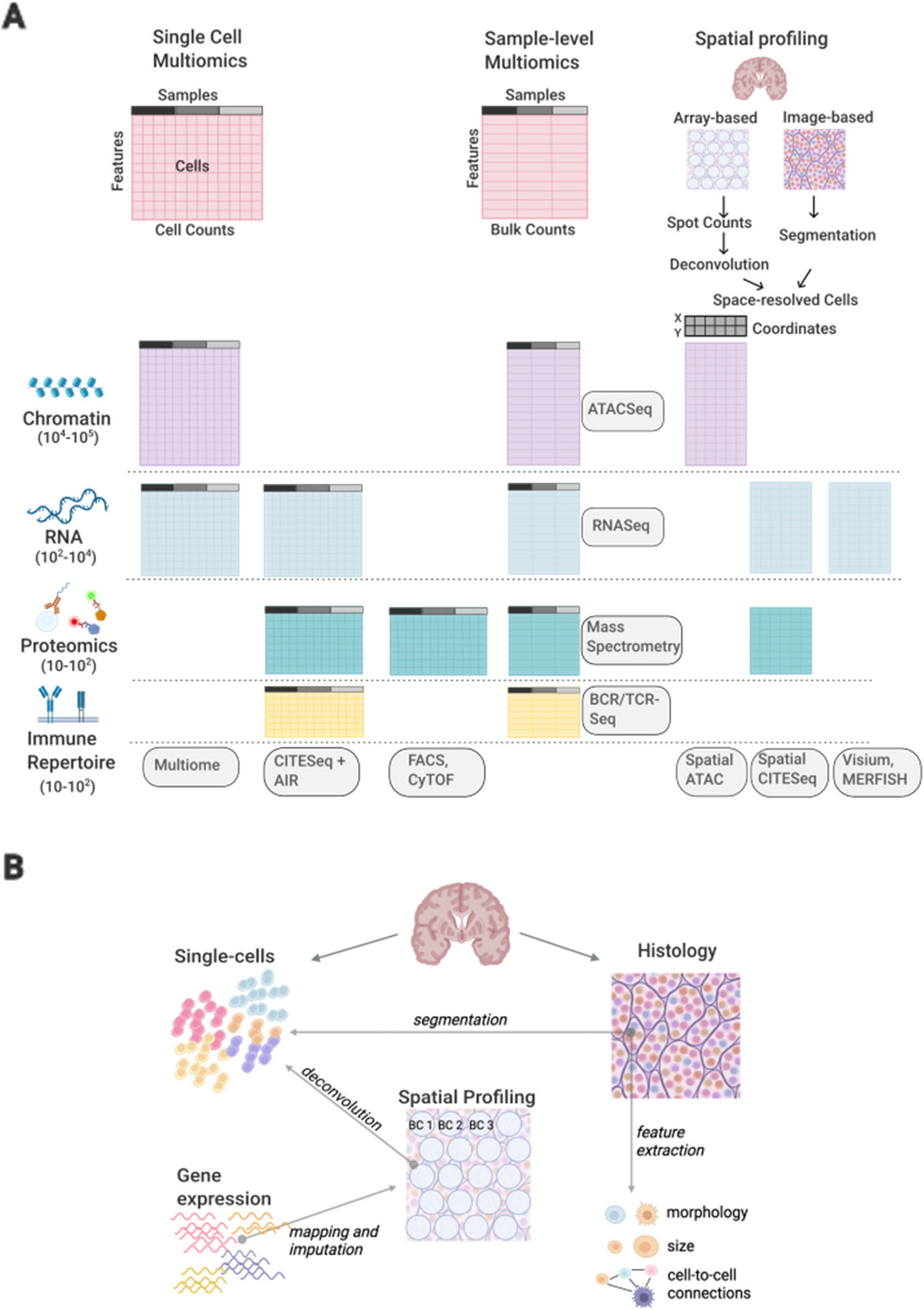
We cloned a 9-residue hemagglutinin (HA) tag into the extracellular domain of KCNE1 (between residues 33 and 34; Fig. 1A). KCNE1-HA (33–34) resulted in threefold higher cell surface labeling of the IKs channel complex compared to a previously described HA tag located in a predicted alpha helix (Fig. S5A) [36]. Neither IKs peak and tail current densities, nor activation properties were significantly different between KCNE1-WT and KCNE1-HA (Fig. 1D). Human Embryonic Kidney (HEK) 293T landing pad cells expressing KCNE1-HA were labeled with a fluorophore-conjugated anti-HA antibody, visualized by confocal microscopy, and quantified by flow cytometry (Fig. 1E, F). In cells expressing KCNE1-HA only, minimal plasma membrane anti-HA labeling was present, consistent with previous studies [9, 37]. In contrast, cells coexpressing both KCNE1-HA and KCNQ1 demonstrated substantial plasma membrane anti-HA labeling in live cells, and both plasma membrane and intracellular labeling in permeabilized cells (Fig. 1E) [9, 38]. Quantification of cell surface expression by flow cytometry confirmed these observations (Fig. 1F). A minimal but detectable amount of plasma membrane labeling was seen in KCNE1-HA only cells but the addition of KCNQ1 caused a 6.1-fold increase in cell surface expression (Figs. 1F and S2B). As expected, a known loss-of-trafficking variant (L51H) had minimal cell surface expression and a known gating variant (D76N) had near-WT cell surface expression when coexpressed with KCNQ1 (Figs. 1F and S5B) [39].
A comprehensive library of KCNE1 mutationsWe generated a comprehensive variant library of the 129-residue KCNE1 protein associated with random 18-mer barcodes using inverse PCR mutagenesis with degenerate primers (see “Methods,” Fig. S3A) [22]. We linked 80,282 barcodes to 2368 missense, 100 synonymous, and 124 nonsense KCNE1 variants (95.7% of the total possible variants; Fig. S3B). Each variant was represented by a mean of 31 barcodes (Fig. S3C).
Multiplexed assay of KCNE1 cell surface expressionWe coupled antibody labeling of the extracellular HA tag to deep sequencing of the library barcodes to perform a multiplexed assay of KCNE1 cell surface expression in cells engineered to constitutively express KCNQ1 (Fig. 2A). We used a landing pad cell line that integrated a single KCNE1 cDNA construct downstream of a preexisting promoter in each cell, allowing standardized expression of a single protein variant per cell [19]. The cells were then stained with a fluorophore-conjugated anti-HA antibody, and sorted into four groups based on surface labeling of HA. Variant counts in each group were quantified using high-throughput sequencing (see “Methods,” Figs. 2A and S6A). After quality control, we obtained cell surface expression scores (see Supplemental Methods ) for 98 synonymous, 117 nonsense, and 2339 missense variants (94.2% of all possible variants; Fig. 2B). Scores calculated from three individual replicates were highly consistent (Spearman’s ρ = 0.83–0.89 for pairwise comparisons, p < 0.001; Fig. S6B). Cell surface trafficking scores for synonymous variants were normally distributed (0.988–1.024, 95% confidence interval, Fig. 2B,C). Nonsense variant scores were bimodally distributed (mode 1 = 0.02, mode 2 = 0.74). Nonsense variants from residue 1 to 55 were all trafficking-deficient, whereas nonsense variants at or after residue 56 had near-WT or higher than WT cell surface expression (p = 2.5 × 10−20, Wilcoxon test; Fig. 2B,C), indicating that only 55 residues are needed for KCNE1 expression at the cell surface. We refer to early nonsense variants (residues 1–55) and late nonsense variants (residues 56–129) in descriptions of trafficking experiments below and in the figures. All variants at the residue 1 start codon were complete loss-of-trafficking as expected.
Variants were divided into 6 categories: loss-of-trafficking, partial loss-of-trafficking, possible loss-of-trafficking, normal trafficking, possible gain-of-trafficking, or gain-of-trafficking. We identified 105 loss-of-trafficking, 365 partial loss-of-trafficking, and 310 gain-of-trafficking missense variants (Table S5). We also compared trafficking scores to the few literature reports of cell surface trafficking measurements, typically reported as “normal” or “loss-of-trafficking.” 7/9 variants were consistent: 6 variants with normal or near-normal trafficking (T6F, S74L D76A, D76N, Y81C, and W87R) and 1 loss-of-trafficking variant (L51H; Fig. S6C) [39,40,41,42]. Two variants, R98W and G52R, were previously reported to have normal trafficking but had reduced trafficking scores in our assay (R98W: 0.192–0.637, and G52R: − 0.033–0.247, 95% confidence interval) [9]. We conducted individual cell surface expression studies of these variants and found them to have mean trafficking levels of 53.0% (R98W) and 4.5% (G52R; normalized to WT, Fig. S6D), consistent with their partial loss-of-trafficking and loss-of-trafficking MAVE scores. After inclusion of our repeat validation experiments, the MAVE trafficking scores were fully consistent with individually studied variants (Fig. S6E).
Several interesting and previously unreported findings emerged from the cell surface trafficking scores (Fig. 2F). As mentioned above, nonsense variants after residue 55 were still able to traffic to the cell surface. In the extracellular N-terminus, almost all cysteine variants from residue 2 to 33 had increased cell surface expression. Residues 46–59 in the transmembrane helix, likely juxtaposed to the hydrophobic core of the lipid bilayer, were highly intolerant to polar or charged variation. Variants in residues 61–70, interacting with the intracellular hydrophilic edge of the cell membrane, increased cell surface expression. Predicted alpha helices in the intracellular C-terminus (residues 79–114) had multiple residues intolerant to aromatic or larger aliphatic variation.
Fig. 2
Multiplexed assay of KCNE1 cell surface expression. A To conduct the cell surface expression multiplexed assay of KCNE1, we used an HA epitope in KCNE1, constructed a comprehensively mutated barcoded plasmid pool, and integrated the library into the LP-KCNQ1 cell line. Cells were stained with a fluorophore-conjugated anti-HA antibody, sorted cells into 4 bins by surface KCNE1 levels and each pool was deep sequenced. Variant abundance in each sequenced pool was used to calculate a trafficking score for each variant. The diagram shows three example variant plasmids, each denoted by a different color. The three example variants have different levels of KCNE1-HA surface expression (green: loss-of-trafficking, gray: normal trafficking, yellow: high trafficking), resulting in different patterns of sequencing results in each of the 4 sorted cell pools. B Histogram of normalized trafficking scores by functional class showed a unimodal distribution of missense variant scores centered at the median of synonymous variant scores. A smaller peak at the median of early nonsense variant scores and a tail of super-trafficking variant scores was also seen (Dotted lines: mean ± 1.96 SD for the synonymous distribution). C Most nonsense variants (1 per residue) from residues 1–55 (brown) are loss-of-trafficking but nonsense variants after residue 55 (red or pink) are gain-of-trafficking or WT-like. Synonymous variants (1 per residue) are represented in green (Dotted lines: mean ± 1.96 SD for the synonymous distribution). D Trafficking scores in the presence and absence of KCNQ1 were highly correlated with the exception of variants at residues 5, 7, 26, and 28 which disrupted glycosylation sites (highlight). Cysteine variants at these sites did not follow this trend (annotation). See Fig. S5 for a full description of the KCNQ1− trafficking assay. Dotted lines: mean ± 1.96 SD for the synonymous distribution. E Near complete representation of variants was scored at each position in the KCNE1 protein (max = 21). F A heatmap showing trafficking scores. Red (score = 0), white (score = 1), and blue (score = 2) indicate low, WT-like, and high cell surface expression, respectively. Dots in squares indicate WT amino acids at that position. The colored ribbon indicates secondary structure (purple = alpha helices, light blue = transmembrane domain, yellow = unstructured regions). G For each residue, the proportion of gain-of-trafficking (GoT) and loss-of-trafficking (LoT) missense variants is displayed
As HEK293T cells expressing only KCNE1 exhibited detectable levels of anti-HA staining by flow cytometry, we conducted a second MAVE of KCNE1 cell surface expression in the absence of coexpressed KCNQ1 (“KCNQ1− ”; Fig. S7). Trafficking scores for missense variants in the presence and absence of KCNQ1 were highly correlated (Fig. 2D, Spearman’s ρ = 0.88, p < 2 × 10−6), except for variants in the N-glycosylation sites (NXS/NXT motifs) at positions 5–7 and 26–28 [42, 43]. Overall, glycosylation variants comprised 39/50 variants with the largest difference in trafficking scores in the presence and absence of KCNQ1. In the presence of KCNQ1, most glycosylation variants at residues 5 and 7 had normal or near-normal trafficking compared to WT (28/34 normal, possible loss-, or possible gain-of-trafficking; Fig. 2D). However, in the absence of KCNQ1, most glycosylation variants at residues 5 and 7 were partial loss- or possible loss-of-trafficking compared to WT (33/34). On the other hand, in the presence of KCNQ1, almost all glycosylation variants at residues 26 and 28 had gain-of-trafficking scores compared to WT (31/32). However, in the absence of KCNQ1, only N26C and S28C had gain-of-trafficking scores compared to WT, and 27/32 had partial- or possible loss-of-trafficking scores. Thus, most variants disrupting N-glycosylation had higher trafficking scores with coexpression of KCNQ1 when compared to in the absence of KCNQ1, and this increase was especially pronounced at residues 26 and 28 (Fig. 2D). All subsequent mentions of trafficking scores refer to the trafficking assay conducted in the presence of KCNQ1.
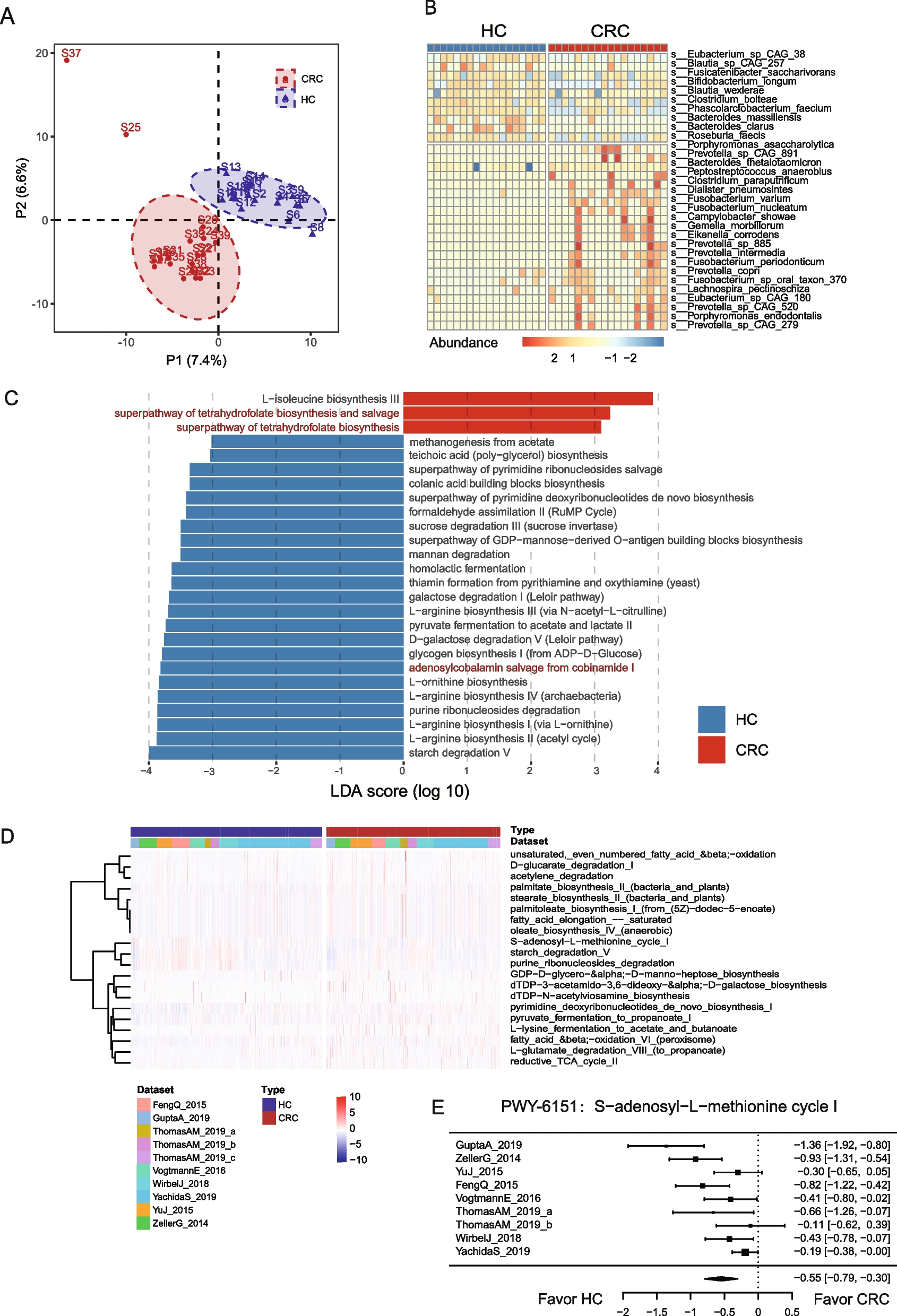
Development of a cell fitness assay for KCNE1 functionSince IKs channels are closed at the resting potential of HEK293T cells (approximately −25 mV) [26], we developed an assay for KCNE1 function by leveraging the electrophysiology of a previously described gain-of-function variant, KCNQ1-S140G [25, 44]. In the presence of KCNE1, KCNQ1-S140G left-shifts the voltage of I Ks channel activation [25] and reduces channel deactivation [24]; in the absence of KCNE1, the channel expresses minimal current (Fig. 3A). These electrophysiological shifts result in substantial KCNE1-dependent K+ efflux at −25 mV. We hypothesized that long-term expression of IKs channels formed by KCNQ1-S140G and normally functioning KCNE1 would increase K+ efflux and thereby decrease cell fitness. To test this hypothesis, we integrated a 1:1 mixture of empty vector control and either wildtype KCNE1-HA or previously studied KCNE1 variants into HEK293T landing pad cells stably expressing KCNQ1-S140G (LP-KCNQ1-S140G, Fig. 3B). We then quantified KCNE1+ and KCNE1− cell proportions over 20 days (Fig. 3C, D). Compared to cells with empty vector controls, cells with WT KCNE1 depleted over time, whereas cells with loss-of-function KCNE1 variants (L51H or D76N) persistently survived. Two previously described KCNE1 gain-of-function variants (G60D and G25V) [7] depleted at a similar rate as KCNE1-HA-WT (Fig. 3D), indicating that this assay is not well-powered to distinguish gain-of-function variants from WT.
Fig. 3
A KCNE1 functional assay using a gain-of-function KCNQ1 variant. A Voltage clamp measurements of outward potassium currents in HEK293T cells transfected with various combinations of WT or S140G KCNQ1 and/or KCNE1-HA. At the resting potential of HEK293T cells (~ −25 mV, dotted line), there is almost no potassium current in cells transfected with KCNQ1 ± KCNE1-HA. However, the gain-of-function KCNQ1-S140G variant results in a leftward shift of voltage of activation and increased current at −25 mV, only in the presence of KCNE1. B Experiment to validate KCNE1 selection assay using LP-KCNQ1-S140G cells transfected with 1:1 pools of KCNE1 variant (pink) and empty vector (red) plasmids. C Representative flow cytometry measurements of cells transfected with 1:1 pools of plasmids. Cells with empty vectors had reduced mCherry fluorescence allowing quantification of distinct cell populations. After 20 days of selection, there was a strong depletion of cells expressing KCNE1-HA-WT but not a loss-of-function variant D76N. D Time course of relative fitness of cells expressing KCNE1 variants compared to cells expressing empty vectors. All values are normalized to the values at day 0. Cells expressing KCNE1-HA-WT or two gain-of-function variants (G25V and G60D) depleted over 20 days but cells expressing two loss-of-function variants (D76N and L51H) persisted in the cell pool. Points and error bars indicate mean and standard error of three replicates
In order to quantify the expression levels of KCNQ1 and KCNE1 in our experiments, we performed RNA sequencing of two stable lines (in 3 replicates each) expressing KCNQ1 (wildtype or S140G), with KCNE1 integrated into the landing pad. We quantified KCNQ1 and KCNE1 expression, measured as fragments per kilobase mapped (FPKM; Table S6). KCNQ1 had a mean FPKM of 629 and 1065 in the wildtype and S140G lines, respectively. KCNE1 had a mean FPKM of 83 and 46, respectively.
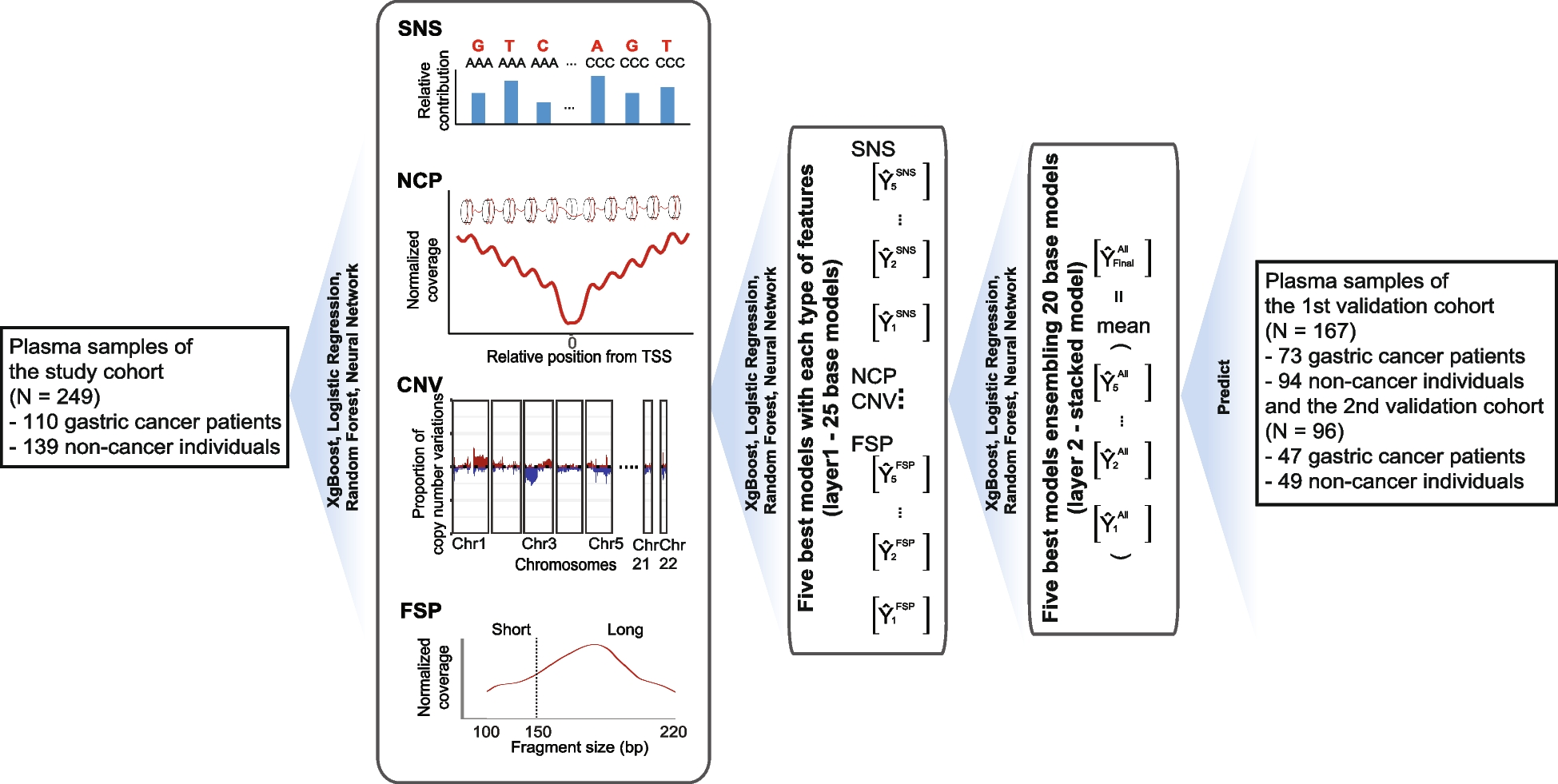
Multiplexed assay of KCNE1 functionWe used this KCNQ1-S140G-based cell fitness assay to conduct a multiplexed assay of KCNE1 function. We hypothesized that KCNE1 variants that decrease I Ks (through multiple mechanisms such as reduced peak current or severe alterations to channel gating) would persist in the cell pool over time, whereas those with normal or increased I Ks would deplete from the pool. To conduct the assay, we integrated the KCNE1 library into HEK293T cells stably expressing KCNQ1-S140G and quantified cell fitness over 20 days (Fig. 4A). After 20 days, cell expressing synonymous variants were depleted and those expressing most nonsense variants persistently survived (Fig. S8A). We also found that depletion of missense and nonsense variants from the cell pool expressing the comprehensive library was only observed against the background of KCNQ1-S140G (Fig. S8B). These changes over time were not seen in the presence or absence of KCNQ1-WT (Fig. S8B), suggesting that selection against cells with functional channels is due to the gain-of-function S140G variant. Variant depletion from day 0 to day 20 was used to calculate normalized KCNE1 functional scores for 98 synonymous, 121 nonsense, and 2320 missense variants (93.7% of all variants; Figs. 4B and S8C). Functional scores across three replicates were highly consistent (Spearman’s ρ = 0.81 for all 3 pairwise comparisons; Fig. S8D). Synonymous variants followed an asymmetric unimodal distribution (median = 0.99, IQR = 0.27, Fig. 4B). Nonsense variants were bimodally distributed (mode 1 = 0.02, mode 2 = 1.08); nonsense variants at residues 1–104 were loss-of-function, whereas nonsense variants at residue 105–129 had WT-like function (p = 2.6 × 10−13, Wilcoxon test, Fig. 4C). Thus, across the trafficking and functional assays, three broad classes of nonsense variants were present based on residue number: #1–55 caused loss of trafficking and function, #56–104 caused WT-like or elevated trafficking but loss-of-function, and #105–129 caused near WT-like trafficking and function (Fig. S9A). All variants at residue 1 were loss-of-function as expected. Missense variant functional scores were bimodally distributed (mode 1 = 0.1, mode 2 = 1.12). The two modes approximately corresponded to the peaks of the early nonsense and synonymous distributions. Variants were divided into 6 categories: loss-of-function, partial loss-of-function, possible loss-of-function, normal function, possible gain-of-function, or gain-of-function. We identified 173 loss-of-function, 401 partial loss-of-function, and 1 gain-of-function missense variants (Table S7).
Fig. 4
Multiplexed assay of KCNE1 function. A Schematic of multiplexed assay of KCNE1 function. A comprehensively mutated barcoded plasmid pool was integrated into LP-KCNQ1-S140G cells. Cells were grown for 20 days and cell pools at 0 days, 8 days, and 20 days were deep sequenced. Different cell colors represent example KCNE1 variants (green: loss-of-function, blue: low function, gray: normal function, yellow: high function). Cells with functional KCNE1 variants were depleted from the cell pool over time. B Histogram of normalized functional scores by functional class showed a bimodal distribution of missense variants (Dotted lines: mean ± 1.96 SD for the synonymous distribution). C Nonsense variants (1 per residue) up to residue 104 are functionally deleterious (brown and red), including residues 56–104 that were dispensable for cell surface trafficking (red). Synonymous variants (1 per residue) are represented in green (Dotted lines: mean ± 1.96 SD for the synonymous distribution). D Relationship between trafficking scores in the presence of KCNQ1 and functional scores for missense variants showed that most deleterious variants disrupt gating (Dotted lines: mean + 1.96SD for the nonsense distribution, and mean ± 1.96 SD for the synonymous distribution). E Near complete representation of missense variants was scored at each position in KCNE1 (max = 21). F KCNE1 functional score heatmap. Red (score = 0), white (score = 1), and blue (score = 2) indicate loss-of-function, normal function, and gain-of-function, respectively. Dots in squares indicate WT amino acids at that position. The colored ribbon indicates secondary structure as in Fig. 2. G For each residue, the proportion of loss-of-function missense variants is displayed
There was a complex and statistically significant relationship between trafficking and functional scores (p < 2.2 × 10−16, chi-square test; Fig. S9B-D and Table S8). 84/105 (80%) loss-of-trafficking variants had partial, possible, or loss-of-function scores (Table S8, Fig. S9D). This includes known LQT5-associated trafficking and function-disrupting variants such as L51H [39]. However, only 103/365 (28.2%) of the partial loss-of-trafficking variants fell into loss-of-function categories. We validated this finding by patch clamping R32T, a partial loss-of-trafficking variant (0.54) with a normal functional score (0.99). R32T had near-normal function by patch clamping (85% of WT peak current and a − 0.1 mV shift in the voltage of half activation; Fig. S10A). Thus, partial loss-of-trafficking KCNE1 variants can have mild impacts on channel function. 422 of 574 loss- or partial loss-of-function variants (73.5%) did not have reduced cell surface expression (Fig. 4D, Table S8). We refer to these as “gating variants.” These included known gating variants, such as D76N [39].
Using the heatmap of KCNE1 functional scores, we examined regions that were variation-intolerant (i.e., regions that had consistently low scores when mutated to non-WT residues). Variation-intolerant regions were located in the transmembrane helix (residues 61–73) and the transmembrane-proximal cytosolic alpha helix (residues 77–82; Fig. 4F). Common polymorphisms (G38S, D85N) had WT-like functional scores, consistent with their minimal effects on baseline QT interval. Residue 85 is otherwise intolerant of variation and D85N is only one of 2 missense variants at this residue with WT-like score estimates (D85N: 0.79–1.04, and D85E: 0.68–0.98, 95% confidence interval). Variants that disrupted glycosylation sites (residues 5–7 and 26–28) increased cell surface expression in the presence of KCNQ1, but displayed normal function in our functional assay; 60/66 of all missense variants at glycosylation sites had normal functional scores, 2/66 were possible gain-of-function, and 4/66 were possible loss of function (Fig. S9E and F). This appears to be consistent with previous work showing that manipulations to the glycosylation sites have dramatic effects on glycosylation but minimal effects on channel function [42, 43]. Similarly, N-terminal cysteine residues, while gain-of-trafficking, did not drastically affect function in the functional assay. However, a caveat is that the functional assay is not well-powered to detect gain-of-function variants (Fig. 3). Two representative variants (Y107R and C106L), classified as possible gain-of-function in the functional assay, were studied by patch clamp and had gain-of-function properties (Fig. S10B) [42, 43].
The coding region of KCNE1 is completely contained within a single exon, 50 bp from the nearest splice site. Nevertheless, we considered potential effects of KCNE1 variants on RNA splicing using the machine learning algorithm SpliceAI (Supplemental file 3) [34]. Of the 1109 investigated single-nucleotide variants affecting the coding region of KCNE1, no variants had SpliceAI scores above the SpliceAI “recommended” cutoff of 0.5, and only 8 variants had SpliceAI scores above “high recall” threshold of 0.2 [34]. These 5 missense and 3 synonymous variants all had SpliceAI scores between 0.2 and 0.4, and thus are moderately predicted to disrupt splicing. One of the candidate splice-disrupting missense variants also had a low functional score (L59Q, estimate − 0.09–0.11, Fig. S10C).
To determine whether KCNE1-HA and KCNQ1-S140G retain a response to beta-adrenergic stimulation, we measured I Ks response after addition of forskolin and isobutylmethylxanthin (IBMX), two activators of the adenylyl cyclase-PKA pathway. The compounds had an acute effect of increasing I Ks current (Fig. S10D). Therefore, future work could use the framework established in this paper to measure the comprehensive impact of KCNE1 variants on beta-adrenergic response.
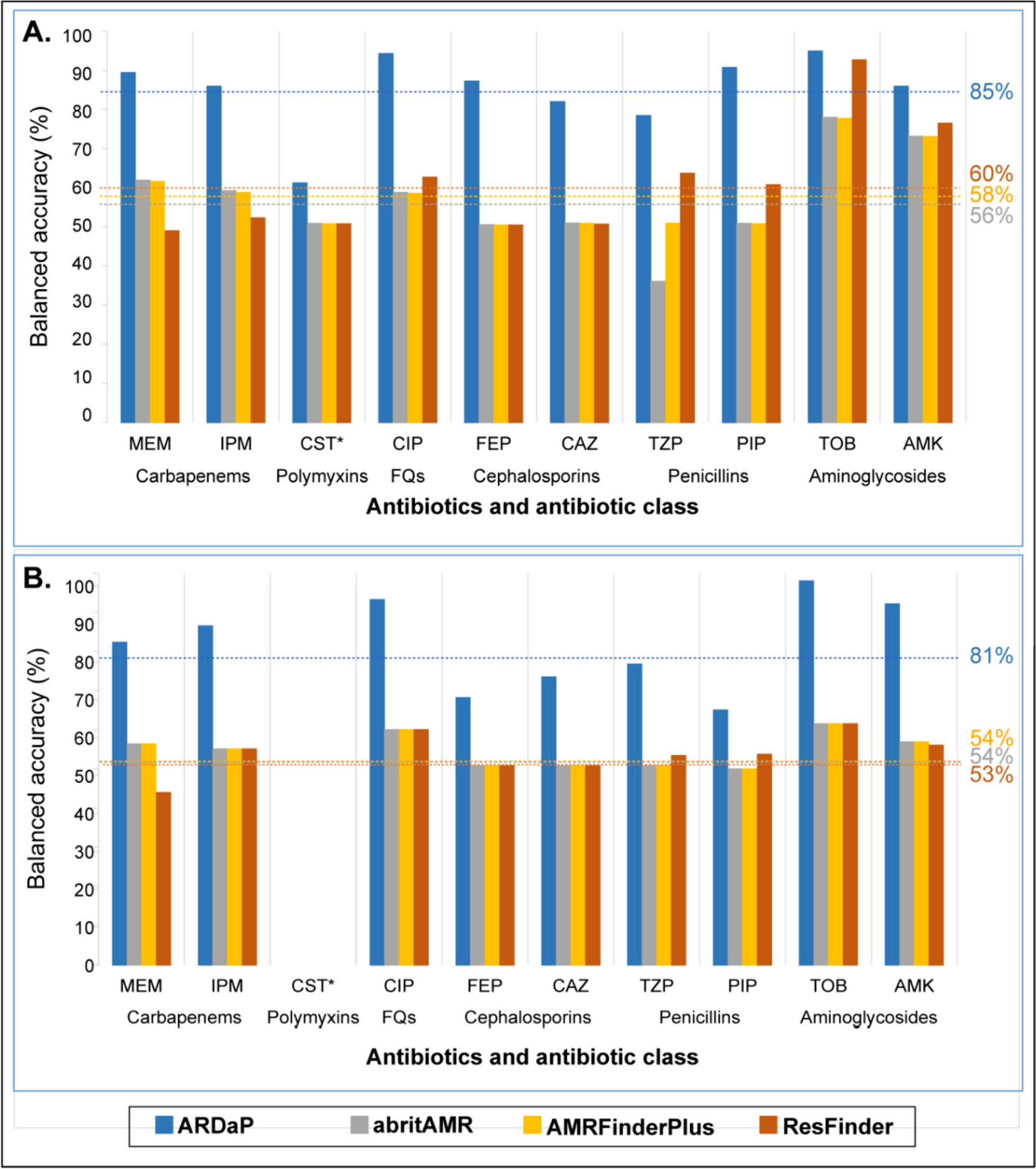
Structural model of the KCNE1/KCNQ1/calmodulin complexAn experimental structure of the KCNE1/KCNQ1/calmodulin complex has not been determined, so we examined multiple AI-based models (Fig. S11). Aligning predicted structures of KCNE1 to a reported cryo-EM structure of the KCNQ1-KCNE3 complex indicated that the conformation obtained from AlphaFold-multimer modeling (see Supplemental Methods) was likely the most biologically meaningful (Figs. 1A and 5). We therefore overlaid mean missense trafficking and missense functional scores on the AlphaFold-multimer structural model of the KCNE1-KCNQ1-calmodulin complex (Fig. 5A–D). As discussed above, the hydrophobic core of the transmembrane helix (residues 44 to 60) did not tolerate polar or charged variation in both trafficking and functional maps. Multiple variants in the internal half of the transmembrane domain (residues 61–70) increased cell surface expression. However, these variants were highly intolerant to variation in the functional assay, highlighting that KCNE1 variants can reduce potassium efflux by disrupting either cell surface expression or interactions with KCNQ1. Given the increased cell surface expression of these variants, we hypothesized that variants in this region likely disrupt normal interactions with KCNQ1 and gating properties of the I Ks channel. Our hypothesis was supported by examining the I Ks channel complex structure, which demonstrated that this region contains extensive contacts with KCNQ1, with 7 of the 9 mutation-intolerant variants within 5 Å of KCNQ1 (Fig. 5F). Polar and charged variation in an additional predicted intracellular helix (residues 85–105 including functionally constrained residues F78, Y81, I82, and W87) preserved cell surface expression. However, the helix was functionally intolerant to variation likely due to observed interaction with calmodulin (Fig. 5G) that affects gating properties of the I Ks complex. We identified 30 KCNE1 residues highly intoleract of variation (defined as positions where > 70% of missense variants were loss- or partial loss-of-function). Of these 30 residues, 22 (73%) were in close contact (< 5 Å) with KCNQ1 (14 residues) and/or calmodulin (9 residues; residue 77 was in close contact with both proteins). These variation-intolerant residues comprise 40% of 55 KCNE1 residues in close contact with KCNQ1 and/or calmodulin. On the other hand, none of the 70 residues where > 50% of the missense variants had WT-like functional scores were in close contact with either KCNQ1 or calmodulin.
Fig. 5
Structural impact of variant effects. A–D KCNE1 structural model, coded by mean missense trafficking scores (A,B) or missense functional scores (C,D) at each residue on the same color scale as in Figs. 2F and 4F (Red = loss-of-trafficking/function, white = WT-like, blue = gain-of-trafficking/function). Dotted lines indicate the approximate location of the plasma membrane. (g) represents glycosylation sites. Panels B and D show KCNE1 in complex with KCNQ1 (green) and calmodulin (gray). E I Ks complex showing locations of panels F and G. F Constrained KCNE1 residues (pink) that make contacts with KCNQ1. KCNE1 residue M62 forms a hydrophobic cluster with KCNQ1 residues F123, F127, and V241, whereas KCNE1 residues Y65 and S68 make polar contacts with KCNQ1 residues D242 and Q260, respectively. G Constrained KCNE1 residues (pink; F78, Y81, I82, W87) that make contacts with calmodulin. Residues on calmodulin not labeled. A PDB model and and Pymol session files with trafficking and functional scores overlaid on the structure are in Supplemental files 7–8
Correlation of MAVE scores with patch clamp and patient dataWe curated a list of 149 KCNE1 variants with previously published patch clamp, trafficking and/or patient data (Supplemental file 2), and conducted additional patch clamp studies of 18 variants (Table S9). The MAVE functional scores were strongly correlated with measured or reported peak currents (ρ = 0.64, p = 2.3 × 10−9, n = 71; Fig. 6A). Thirty nine variants with available patch clamp data were partial loss-of-function or loss-of-function in the MAVE assay (functional scores below the cutoff of 0.44). Thirty three of the 39 variants (85%) had a 50% or greater reduction in peak current compared to wildtype (Fig. 6A). Since the patch clamp data was generated with coexpression of WT KCNQ1, these results suggest that most KCNE1 loss-of-function variants are loss-of-function when coexpressed with either WT or S140G KCNQ1. Although functional scores correlated best with peak current, KCNE1 variants with large shifts in the voltage or kinetics of activation or deactivation were also more likely to have lower scores (Figs. 6B, S12A, and B). We also saw a correlation between functional scores and current measured at −20 mV (ρ = 0.65, p = 6.4 × 10−5; Fig. S12C), though there were fewer variants for this parameter. As expected, the trafficking scores had weaker correlations with patch clamp parameters (Fig. S12D, E, and F). Since transition mutations occur more frequently than transversion mutations, we also investigated the functional effects of transition vs. transversion missense variants. Single-nucleotide variants (SNVs) generated by transversion mutations were more likely to be functionally deleterious compared to variants generated by transition mutations (p = 3.4 × 10−5, Wilcoxon test; Figs. S12H and I). In addition, we hypothesized that functional scores of variants generated by two or three SNVs would be more likely to have altered scores in our functional assay than those generated by one SNV. Indeed, variants generated by more than one SNV tended to have lower functional and trafficking scores (p = 2.0 × 10−2 and 3.4 × 10−5, respectively, Wilcoxon test; Figs. 6B and S12G). These results are consistent with previous calculations showing that the genetic code is optimized to maximize the chemical similarity of amino acids introduced by transition and single-variant mutations [45].
Fig. 6
Functional scores correlate with in vitro assays and clinical outcomes. A Correlation of function scores with peak current (normalized to WT) from patch clamp studies. Blue: currents obtained from literature review, pink: mean of currents from literature review and this study, yellow: currents measured in this study. B Variants generated by single SNVs (blue) are less likely to have low functional scores compared to all other variants (black; p = 2.0 × 10−2, Wilcoxon Test). C Variants present in gnomAD (black) were more likely to have WT-like scores than variants absent from gnomAD (gray; p = 3.9. × 10−3, Wilcoxon test). Mean and confidence intervals are generated by re-sampling both distributions 100 times. D Functional scores by ClinVar classification. E Functional scores for presumed benign and presumed pathogenic variants. See Supplemental file 2 for the literature review
留言 (0)