
記住我



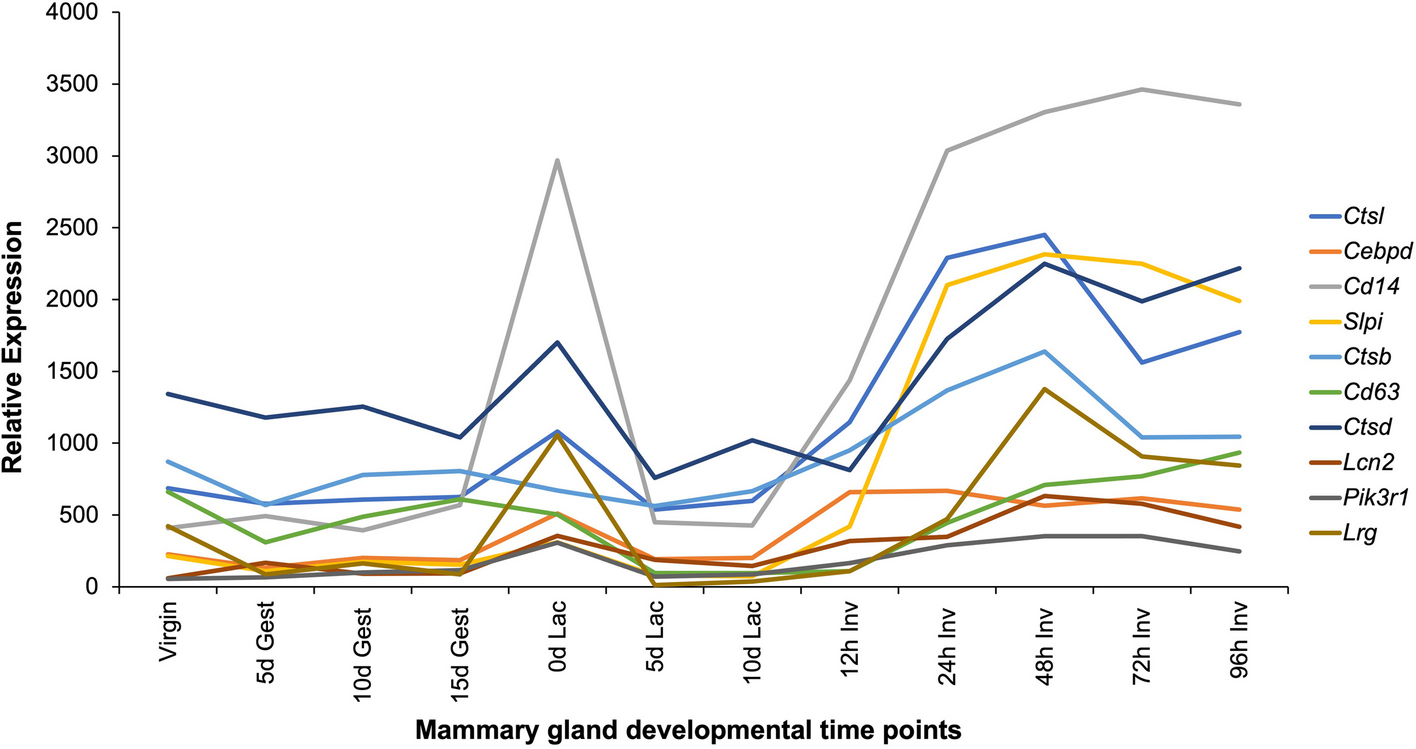

The following protocols describe in vitro biochemical and microscopy-based approaches for investigating the role of STAT3 in regulating lysosomal biogenesis and function in the EpH4 mouse mammary epithelial cell line [20], which may be readily adapted for different cell lines responsive to cytokine-induced STAT3 activation.
Cell culture and OSM stimulation of STAT3 activity in EpH4 cellsThe EpH4 normal murine mammary epithelial cell line was originally derived from the mammary gland of a mid-pregnant BALB/c female mouse [20]. OSM stimulation of EpH4 cells results in STAT3 activation and cell death, mimicking STAT3 dependent LM-PCD of mammary epithelial cells during in vivo mammary gland involution [4, 8, 15, 16, 18]. Thus, this cell line serves as a useful in vitro model for investigating the role of STAT3 signalling in lysosomal function and LM-PCD.
Cell maintenance and passage 1.Maintain EpH4 cells in 10% (v/v) FCS in DMEM in cell culture flasks at 37 °C with 5% CO2. If preferred, 100 U/ml Penicillin and 100 U/ml streptomycin (v/v) may be added to the medium.
2.When cells reach confluency, wash twice with PBS, and incubate with Trypsin–EDTA at 37 °C, 5% CO2 until cells detach.
3.Add 4 volumes of 10% (v/v) FCS in DMEM to inactivate the trypsin.
4.Dilute cell suspension 1:10 – 1:20 in new flasks containing fresh media (e.g. if the total volume of the cell suspension is 10 ml, transfer 1 ml (1:10 split)) into a new flask and top up with fresh medium. Incubate at 37 °C with 5% CO2.
5.Passage cells following steps 2–4 when confluent. Typically, when cells are split into new flasks as a ratio of 1:10 – 1:20 they will require passaging 2–3 times a week.
Stimulation of STAT3 activity in EpH4 cells 1.Trypsinise cells as described above.
2.Seed cells in multi-well plates or cell culture dishes as required for specific assays. For assays performed in standard 6-well tissue culture plates, seed EpH4 cells at a density of 1.0 × 105 cells/well. Incubate at 37 °C with 5% CO2.
3.Upon reaching 50% confluency (typically next day when seeding 1.0 × 105 cells/well in a 6-well plate), stimulate cells with 25 ng/ml of OSM (or 0.1% BSA in PBS as a ‘vehicle’ control) in 1% (v/v) FCS in DMEM (see Note 5).
4.48 h later, renew medium with fresh OSM in 1% (v/v) FCS in DMEM.
5.Analyse cells at different time points as necessary. We recommend analysing EpH4 cells 48—72 h after OSM stimulation, when cells are beginning to undergo programmed cell death, but enough remain for analysis. Immunoblotting with a phospho-STAT3 antibody may be used to validate STAT3 activation in response to OSM stimulation.
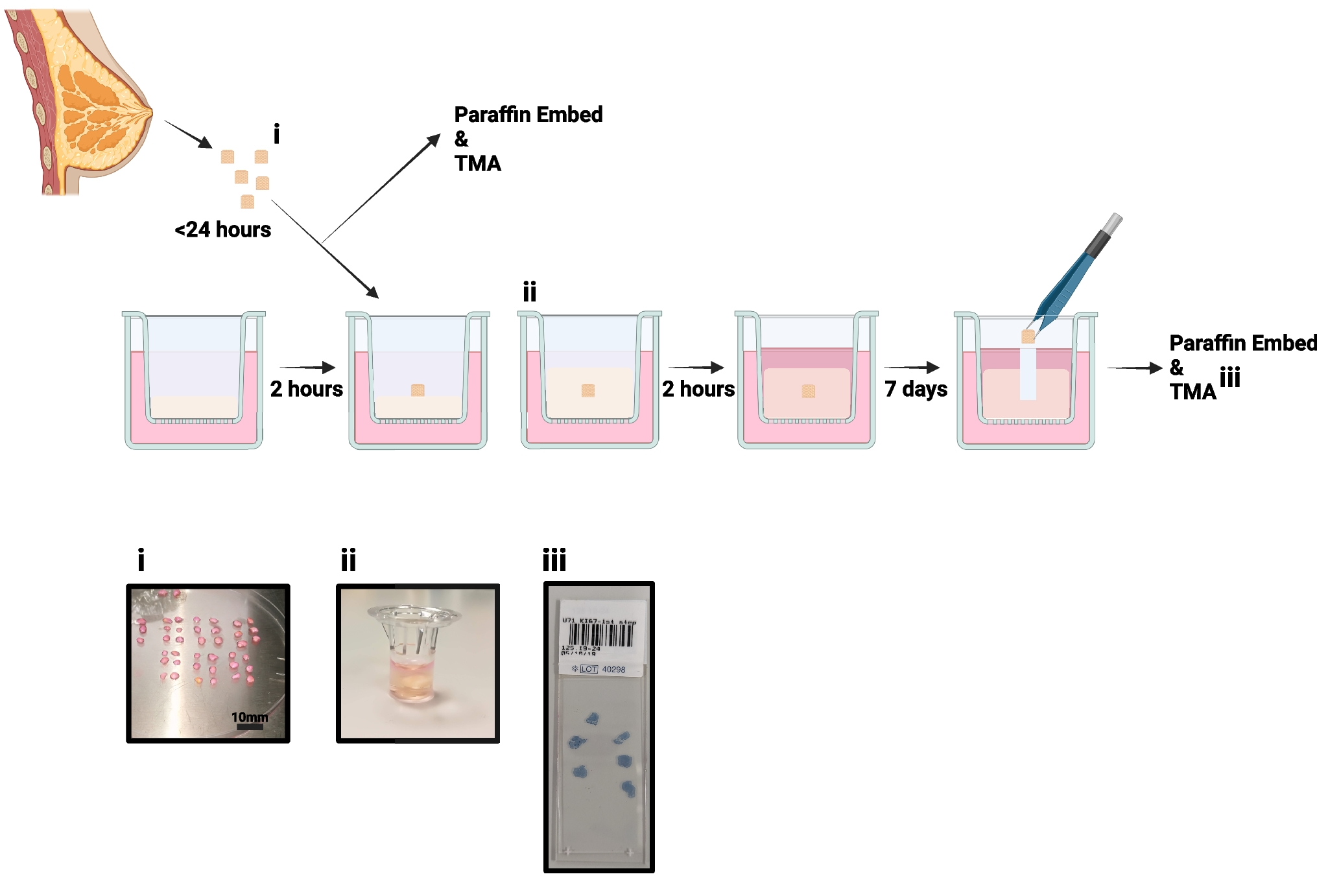
Lysosome isolation using magnetic iron nanoparticlesConventional methods for isolating lysosomes revolve around using density centrifugation, often resulting in preparations contaminated by other organelles (e.g. mitochondria) of a similar density. Moreover, as lysosomes are heterogeneous in nature, variation in lysosomal buoyant density results in their distribution across a wide density gradient, hampering their purification using density centrifugation approaches. To overcome these difficulties, magnetic iron nanoparticles can be used. These nanoparticles are internalised by cells and delivered to lysosomes via the endocytic pathway, enabling their isolation by magnetic chromatography [8, 15, 21,22,23]. We have previously used this approach to isolate lysosomes from EpH4 cells to investigate the impact of OSM-induced STAT3 activation on lysosomal composition and permeability [8, 15]. Lysosomes isolated using magnetic iron nanoparticles can also be analysed by mass spectrometry, or by TEM [15]. This section describes the procedure for isolating lysosomes from EpH4 mammary epithelial cells using Ferrofluid iron nanoparticles (illustrated in Fig. 2), which can also be applied to other cell lines of interest.
Fig. 2
Isolation of EpH4 cell lysosomes using magnetic iron nanoparticles. Schematic representation of the magnetic iron nanoparticle lysosomal purification and fractionation protocol. Adapted from Lloyd-Lewis et al. (2018) [15]. Conditions have been optimised for isolating lysosomes from EpH4 cells to investigate the impact of STAT3 activation (via OSM stimulation) on the lysosomal compartment, which may be optimised or modified for different cell lines. For example, the efficiency of iron nanoparticle uptake can be cell-type dependent, and some cells may require a longer incubation time (provided cells remain healthy with longer iron exposures) or require more starting material to increase the yield of lysosomes isolated for downstream analysis. Isolated lysosomes can be analysed using a variety of downstream assays, including western blotting (illustrated), lysosomal enzyme activity assays or in lysosome leakiness assays. Alternatively, isolated lysosomes can be analysed by mass spectrometry or TEM
Ferrofluid labelling of EpH4 cell lysosomes 1.Seed EpH4 cells at a density of 1 × 106 cells in 15 cm cell culture dishes in 10% (v/v) FCS in DMEM. If not stimulating EpH4 cells with OSM, seed cells at a density of 3 × 106 cells in 15 cm cell culture dishes and proceed directly to step 3 the next day (see Note 6).
2.Next day, treat cells with 25 ng/ml of OSM to stimulate STAT3 activity (as described above).
3.After 48 h of OSM stimulation, prepare the magnetic iron nanoparticles (Ferrofluid) by diluting 1:10 in 10 mg/ml BSA in PBS.
4.Sonicate the ferrofluid-BSA solution 3 times for 15 s on ice to disperse the iron nanoparticles. Rest samples for 15 s on ice between each sonication.
5.Filter-sterilise ferrofluid-BSA solution through a 0.2 µm filter attached to a syringe. Dilute the filter-sterilised ferrofluid-BSA solution 1:10 in 1% (v/v) FCS in DMEM.
6.Replace the media in the 15 cm dishes with the ferrofluid-containing DMEM and incubate for 4 h (‘label’ period).
7.Subsequently, wash EpH4 cells 3 times in PBS, and replace media with fresh 1% (v/v) FCS in DMEM and 25 ng/ml of OSM (or 0.1% (w/v) BSA in PBS as a ‘vehicle’ control) for a further 20 h (‘chase’ period).
Magnetic isolation of iron nanoparticle-containing lysosomes 8.Following the chase period (OSM stimulation time of 72 h), wash EpH4 cells 2 times in PBS.
9.Scrape cells in 1 ml of cold PBS and transfer to 1.5 ml microcentrifuge tubes.
10.Centrifuge at 900 × g for 3 min at 4 °C. Remove supernatant.
11.Add 700 µl of subcellular fractionation buffer containing fresh cOmplete Protease Inhibitor Cocktail (see Note 2) to cell pellets. Homogenise cells in a tight-fitting Dounce homogenizer with five strokes.
12.To remove nuclei and debris, spin the homogenate at 800 × g for 10 min at 4 °C. Transfer the supernatant to a clean 1.5 ml microcentrifuge tube and spin again at 800 × g for 10 min at 4 °C to ensure complete removal of contaminating heavy membranes.
13.Transfer the resulting supernatant (PNS; post nuclear supernatant) to a clean 1.5 ml microcentrifuge tube and load onto a magnetic rack (retain 50 µl for analysis by immunoblotting if required). Incubate for 1 h at 4 °C on a rocker.
14.Following incubation, leave tubes on the magnet while removing the supernatant (abbreviated as S). Retain 50 µl for analysis.
15.Wash tubes attached to the magnetic rack 3 times with 1 ml of cold subcellular fractionation buffer. After the last wash, remove tubes from the magnetic rack and spin at 14,000 × g for 15 min at 4 °C to pellet magnetite-containing lysosomes.
16.Resuspend and combine pellets in a final volume of 200 µl of subcellular fractionation buffer and transfer to a new tube (‘whole lysosomes’) to remove any contaminating proteins that may have bound non-specifically to the side of the original tube. Pellet whole lysosomes, PNS and S samples by centrifuging at 14,000 × g for 15 min at 4 °C.
17.To extract protein lysates from the PNS and S samples, add 50 µl of modified RIPA buffer to pelleted material and incubate for at least 10 min on ice. To obtain “total” lysosomal protein lysates, incubate the pelleted ‘whole lysosomes’ fraction in 0.1% (v/v) Triton X-100 in PBS for 10 min on ice, intermittently vortexing every 2.5 min.
18.Subsequently spin all samples at 17,000 × g for 10 min at 4 °C and transfer the supernatant into clean 1.5 ml microcentrifuge tubes for downstream analysis (e.g. by immunoblotting, lysosomal leakiness assays as described in the subsequent section on measuring lysosomal leakiness). Alternatively, to separate lysosomal content and membrane fractions, continue as described below for fractionation of purified lysosomes.
Fractionation of purified lysosomes (optional) 19.If necessary to separate the lysosomal content from the lysosomal membrane fraction, resuspend the ‘whole lysosome’ fraction (isolated in Step 16) in lysosome membrane fractionation buffer and incubate on ice for 30 min. Alternatively, to improve lysosomal rupture, freeze samples in liquid nitrogen and thaw at 37 °C five times.
20.Pellet lysosomal membranes by spinning at 15,000 × g for 30 min at 4 °C.
21.Transfer the supernatant (lysosomal content (LC) fraction) to a new tube.
22.Resuspend the remaining pellet in 50 μl of 0.1% (v/v) Triton X-100 in PBS, incubate for 10 min on ice, intermittently vortexing four times.
23.Centrifuge tubes at 17,000 × g for 10 min at 4 °C. Transfer the supernatant (lysosomal membrane (LM) fraction) to fresh tubes.
24.Lysates obtained from total lysosome fractions (obtained in Step 17 in ‘Magnetic isolation of iron nanoparticle-containing lysosomes’ protocol) or LC and LM sub-fractions can be further analysed by immunoblotting or mass spectrometry analyses [15] (see Note 7).
Measuring lysosomal leakiness in response to STAT3 activationOSM treatment of EpH4 cells results in lysosomal membrane permeabilisation, leading to the leakage of lysosomal hydrolases into the cytosol [4, 8]. To measure the degree of lysosomal leakage, the activity of lysosomal cysteine cathepsins in the cytosolic compartment of EpH4 cells can be assessed using a sensitive enzyme activity assay [18, 24]. Cytosolic preparations can be obtained using a buffer containing the glycosidic detergent digitonin at a concentration that enables the extraction of cytosolic proteins from EpH4 cells without damaging lysosomal membranes. As OSM treatment also increases cathepsin protein expression in EpH4 cells, cathepsin activity in the cytosolic extraction must be normalised to total activity, which can be measured in total cell extracts obtained using 0.1% (v/v) Triton X-100 in PBS. Subsequently, a kinetic assay based on the cleavage of the fluorescent molecule AMC from the synthetic substrate Z-Phe-Arg-AMC by lysosomal cysteine cathepsins can be used to assess cytosolic cathepsin activity over time. The initial rate of fluorescence (corresponding to the initial rate of cathepsin activity (fluorescence/min)) is subsequently determined from the linear part of the resulting curve [18, 24]. Alternatively, lysosomes can be isolated directly using the purification method described above for in vitro leakage assays.
Method 1: Assessing lysosomal leakiness by measuring cytosolic cathepsin activity 1.Seed EpH4 cells at a density of 1.0 × 105 cells/well in a 6-well tissue culture plate in 10% (v/v) FCS in DMEM. Next day, stimulate STAT3 activity using OSM as described above.
2.At the desired timepoint after OSM stimulation, remove media and wash cells in PBS. Incubate cells in Trypsin–EDTA until detached. Inactivate trypsin in 10% (v/v) FCS in DMEM and transfer samples to 15 ml centrifuge tubes.
3.Centrifuge samples at 200 × g for 5 min. Resuspend cell pellets in 1–2 ml of DMEM.
4.Count cells using a haemocytometer. Transfer 175,000 cells into two 1.5 ml centrifuge tubes per condition.
5.Centrifuge samples at 1000 × g for 3 min at 4 °C.
6.For each condition, resuspend cell pellets in 300 μl of fractionation buffer containing 25 μg/ml digitonin (for cytosolic extraction) or 0.1% (v/v) Triton X-100 in PBS (for total cell extraction).
7.Incubate cells on ice for 10 min. Pulse vortex samples every 2.5 min.
8.Centrifuge samples at 16,000 × g for 2 min at 4 °C.
9.Transfer supernatant into clean 1.5 ml tubes. For each condition there should be a digitonin and Triton X-100 extracted sample.
10.Per well of a 96 well plate (3 wells per sample), add 10 μl of the extracted sample, 160 μl of cathepsin reaction buffer and 30 μl of substrate (final concentration 50 μM) in reaction buffer (2 μl of 5 mM Z-Phe-Arg-AMC in 28 μl of cathepsin reaction buffer). In addition, prepare 3 wells of 200 μl of cathepsin reaction buffer only (‘Blank’ control), and 170 μl of cathepsin reaction buffer + 30 μl of substrate/reaction buffer (i.e. without sample, ‘Assay control’).
11.Measure fluorescence at 1 min intervals for 1 h at 37 °C in a fluorescent plate reader (excitation: 380 nm, emission: 442 nm).
12.Subtract background (mean of assay control wells) from all measurements and plot fluorescence over time for each sample. From the linear part of the resulting curve determine the initial rate of fluorescence for each sample corresponding to the initial rate of cathepsin activity (fluorescence/min).
13.Normalize cytosolic activity to total activity (measured in samples extracted with 0.1% (v/v) Triton X-100 in PBS). Typically, a 1.5-fold increase in cytosolic cathepsin activity is observed in EpH4 cells treated with OSM.
Method 2: Assessing lysosomal leakiness in isolated EpH4 cell lysosomes 1.Isolate lysosomes from EpH4 cells stimulated with OSM to induce STAT3 activity (as described above).
2.After pelleting magnetite-containing lysosomes (Step 16 in ‘Magnetic isolation of iron nanoparticle-containing lysosomes’ protocol), resuspend in 200 µl of subcellular fractionation buffer and aliquot equal amounts (30 µl) into individual tubes (e.g. 4 tubes per treatment condition representing 0, 30, 60 and 90 min timepoints as depicted in Fig. 3).
3.Take one tube (per treatment condition) and centrifuge immediately at 14,000 × g for 15 min at 4 °C (time = 0 min). Carefully remove the supernatant without disturbing the pellet and snap freeze both the collected supernatant (S0) and pellet (P0). These fractions indicate the level of background leakage that occurs during processing.
4.Incubate other tubes at 37 °C with agitation for 30, 60, and 90 min respectively.
5.At the defined timepoints, centrifuge tubes at 14,000 × g for 15 min at 4 °C and carefully remove the supernatant without disturbing the pellet. Snap freeze both the collected supernatant (S 30/60/90) and pellet (P 30/60/90).
6.Resuspend the pellets (P fraction) in 150 μl of RIPA buffer (with protease inhibitors added fresh) and incubate for 30 min on ice with intermittent vortexing (3 pulses of 10 s). Centrifuge at 16,000 × g at 4 °C, for 15 min and remove the supernatant. S fractions from step 5 do not require further lysis.
7.For immunoblotting analysis, load equal volumes of the supernatant/pellet fractions per condition.
8.Probe western blots with cathepsin B and L antibodies to visualise the degree of leakiness of cathepsin hydrolases into the supernatant fraction under different treatment conditions (i.e. vehicle and OSM stimulated conditions). Immunoblotting with a LAMP2 antibody can be used to validate lysosomal integrity during the assay, in addition to standardising the amount of lysosomal material in a pellet during the assay.
Fig. 3
Schematic overview of the lysosomal leakiness assay. The permeability of isolated lysosomes can be assessed by western blotting for cathepsin B and L antibodies to visualise the degree of leakiness of cathepsin hydrolases into the supernatant fraction under different treatment conditions (i.e. vehicle and OSM stimulated conditions in EpH4 cells). Immunoblotting for LAMP2, a lysosomal membrane protein, can be used to standardise the amount of lysosomal material in a pellet during the assay. Alternatively, cathepsin activity levels can be assessed in supernatant and pellet fractions using enzyme activity assays
CRISPR/Cas9 mediated deletion of STAT3 in cell linesThe CRISPR/Cas9 system from Streptococcus pyogenes is the most widely adopted genome editing technology in use today due to its ease of use, high efficiency, and versatility of formats. Like many cell lines, EpH4 cells are highly refractory to transfection using lipofection reagents, but are readily transduced with lentivirus, which we implemented for CRISPR reagent delivery. We chose to use pLentiCRISPRv2 as it provides an all-in-one Cas9 and single guide RNA (sgRNA) expression system and allows for positive selection of transduced cells using puromycin. When a high efficiency guide is selected, the resulting pool of cells are often virtually a complete knockout, with no protein detectable by western blot. This is particularly important for epithelial cell lines such as EpH4 cells, where single cell cloning is best avoided as derived clones are likely to possess an altered phenotype from adapting to growth without normal epithelial cell-to-cell attachment and signalling. As the pool of cells transduced with pLentiCRISPRv2 constitutively express Cas9 and the sgRNA, the potential for cutting the genomic DNA at unintended loci (off-target sites) is increased. Therefore, a number of controls are essential when using all-in-one CRISPR lentivirus: 1) at least 2 independent pools of knockout cells should be established using different guide sequences that target the same gene (as any off-targeting cutting sites will not be shared); 2) a pool of cells targeted with a guide that does not have any matches in the genome (non-targeting guide) to control for any effects of lentiviral transduction, Cas9 expression and puromycin selection; and 3) a non-transduced “wild-type” control pool. To minimise the time for off-target cutting to accumulate, transduced cells should be cultured for the shortest number of passages as possible post-transduction (see Note 8). When designing guide sequences to target the gene of interest, use a tool such as that published by Doench et al. [25] to select for guides with a predicted high cutting efficiency (high on-target score) and low chance of cutting at other sites (low off-target score). Suggested guides for targeting mouse STAT3 and a non-targeting control guide are provided in Table 1.
Table 1 Primer sequences for ‘CRISPR/Cas9 mediated deletion of STAT3 in cell lines’ protocolCloning CRISPR guides into pLentiCRISPRv2 1.Prepare a batch of pLentiCRISPRv2 plasmid cut with the restriction enzyme BsmBI. Digest 2–3 µg of plasmid with 10 U of enzyme for 3 h at 55 °C. Gel extract the larger 12 kb fragment (using any commercial gel extraction spin column kit), avoiding the 2 kb filler sequence fragment. Quantify DNA concentration using a Nanodrop.
2.Phosphorylate and anneal the forward and reverse guide oligos by setting up the following 10 µl reaction: 1 µl of each oligo (each at 100 µM initial concentration), 6.5 µl of nuclease free water, 1 µl of 10X T4 ligase buffer and 0.5 µl of T4 DNA polynucleotide kinase. Place the reaction in a thermocycler and run the following cycle: 37 °C for 30 min, 95 °C for 5 min, then ramp down as slowly as possible (usually 5 °C per min) to 20 °C. Dilute the annealed oligos 1:200 in nuclease free water.
3.Ligate the annealed oligos into the cut plasmid in the following reaction: cut plasmid (20–50 ng), diluted annealed oligos (1 µl), 10X T4 DNA ligase buffer (1 µl), water to 9.5 µl and 0.5 µl of T4 DNA ligase added last. Prepare a control ligation with no annealed oligos (vector alone control). Incubate the ligation at room temperature for at least 1 h.
4.Transform a chemically competent RecA negative E.coli strain such as Stbl3 or NEBstable according to manufactures’ instructions. Spread on LB agar plates containing 100 µg/ml ampicillin and culture for 24 h at 30 °C. There should be at least tenfold more colonies on the vector + oligos plate than the vector alone plate.
5.Pick 2 colonies per guide, grow an overnight culture in LB containing 100 µg/ml of ampicillin for 16 h at 30 °C in a shaking incubator at approximately 250 r.p.m.
6.Prepare miniprep DNA using a standard spin-column based kit, and sequence using the hU6-fwd primer from Table 1.
7.For colonies containing a correct guide sequence, prepare a larger scale plasmid prep (midi or maxiprep) to obtain sufficient DNA for lentivirus packaging (at least 20 µg).
Packaging lentivirus for transduction (see Note 9) 1.Day 1:
For each lentivirus to package, seed 1.5 x 106 HEK293T cells in one 10 cm dish containing 10 ml 10% (v/v) FCS in DMEM. Culture cells at 37°C overnight.
2.Day 2:
(a)In a 15 ml tube, prepare the plasmid transfection solution by mixing 18.25 µg of pLentiCRISPRv2 + guide plasmid, 5.84 µg of pMD2.G plasmid and 11.68 µg of pCMVΔR8.91 plasmid in 1.5 ml of HBS. Add 82 µl of PEI dropwise, then vortex vigorously for at least 10 s.
(b)Incubate at room temperature for 10 min.
(c)Add 4 ml of room temperature OptiMEM and mix by inverting the tube 6–8 times.
(d)Incubate for a further 10 min at room temperature.
(e)Aspirate media from cells and add the ~ 5.5 ml of transfection mixture and return to the incubator for 4 h.
(f)Aspirate the transfection mixture and replace with 8 ml of 10% (v/v) FCS in DMEM and culture (see Note 9).
3.Day 4:
(a)48 h after transfection, collect the culture supernatant (containing lentivirus) from the HEK293T cells using a 10 ml syringe. Filter through a 0.45 µm filter into a 10 ml tube. This is the 48 h viral supernatant; use for transduction immediately (following protocol below), or store at -80 °C until required.
(b)Add 8 ml of 10% (v/v) FCS in DMEM to the HEK293T cells and culture for a further 24 h to collect additional lentivirus.
4.Day 5: Collect the culture supernatant (containing lentivirus) from the HEK293T cells using a 10 ml syringe. Filter through a 0.45 µm filter into a 10 ml tube. This is the 72 h viral supernatant; use for transduction immediately (using protocol below) or store at -80 °C until required.
Transducing, selection, and cryopreservation of EpH4 cells (see Note 9) 1.The day before viral transduction, seed 1.0 × 105 EpH4 cells per well of a 6-well tissue culture plate in 2 ml of 10% (v/v) FCS in DMEM. Include one extra well as a control for puromycin selection. Culture cells overnight.
2.Add polybrene at a final concentration of 4 µg/ml to the 48 h viral supernatant and mix well. 2 wells of a 6-well plate can be transduced with the viral supernatant from one 10 cm dish.
3.Aspirate media from EpH4 cells and add the viral supernatant/polybrene mixture. Culture cells overnight.
4.Next day, repeat steps 2–3 using the 72 h viral supernatant to increase efficiency of lentiviral transduction.
5.Transduced EpH4 cells will likely be confluent at this stage, so passage the cells as described above. Maintain two wells of a 6 well plate per transduction and one control well.
6.24 h after passaging, add puromycin to a final concentration of approximately 1 µg/ml (optimal concentration should be determined empirically for each batch of puromycin and specific cell type).
7.Monitor the plate for cell death by observing cell detachment from the dish under a phase contrast microscope. When all the cells in the control well are dead (after approximately 48–72 h), the transduced cells should be > 90% viable, and confluent. Passage the cells, combining the 2 wells into a T175 tissue culture flask.
8.48 h later, the flask should be 80–90% confluent. Cryopreserve several vials of cells for later use by trypsinising the cells, pelleting at 300 × g for 5 min, then resuspending in 10% (v/v) DMSO and 10% (v/v) FCS in DMEM at a concentration of 1–2 × 106 cells per ml. Add 1 ml per cryovial, place inside a Mr. Frosty freezing container and leave at -80 °C overnight before transferring to liquid nitrogen for long-term storage. These cells are 3 passages post-transduction (see Note 8).
Assessing STAT3 deletion at the genomic DNA level using TIDE analysis 1.Prepare genomic DNA from 0.5–1.0 × 106 cells from the STAT3 targeted cell lines and a wild-type control line.
2.Perform a PCR using the primers included in Table 1 using standard procedures for a commercial polymerase in a 50 µl reaction. Use 50 ng of genomic DNA as template.
3.Run 5 µl of the PCR product on a 1% (w/v) agarose in TAE gel and confirm the presence of a single product of the expected size (see Table 1).
4.Purify the remaining 45 µl of PCR product using a standard commercial PCR clean-up kit.
5.Submit samples for Sanger sequencing. Sequence with both the forward and reverse primer (Table 1).
6.Analyse the sequencing results using the TIDE web interface (http://shinyapps.datacurators.nl/tide/ (see Note 10)).
7.The software will provide a quantification of the level of indels found in the sample, an R2 value for goodness of fit, and the length of the indels. These should be similar for both the forward and reverse sequence for
留言 (0)