
記住我
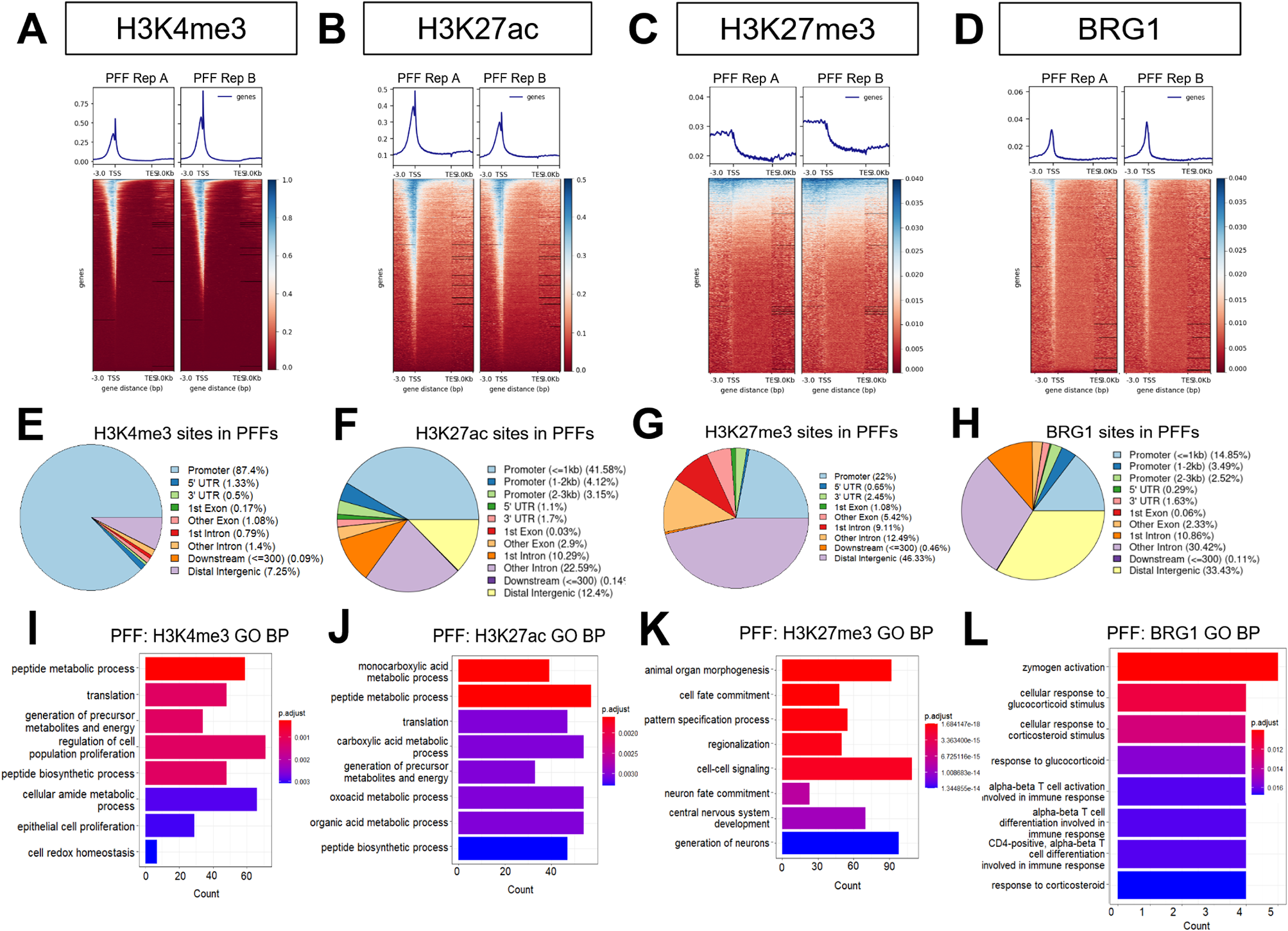

To explore the effects of maternal nicotine exposure on testis morphology of the progeny, we compared hematoxylin and eosin-stained frozen testis sections in 11-week-old male animals born to nicotine-exposed or control mothers. We examined the tubules at stage VI-VII in which all cell types are present to focus on the homogenous presence of the cell types between groups. In rats, there are 14 stages of spermatogenesis that can be distinguished in seminiferous tubules [20]. The various stages are represented by different types and numbers of cells [20]. The spermatogonia and early meiotic cells (leptotene-zygotene stage) are normally found in proximity to the membrane of seminiferous tubule cells, whereas more advanced pachytene and haploid cells are closer to the lumen (Fig. 1A). Stages VI and VII in the rat testis are defined based on the appearance and development of specific germ cells within the seminiferous tubules. Stage VI is characterized by the presence of late spermatids with elongated heads. The acrosome and nuclear shaping are more pronounced, and these spermatids are approaching the final stages of their maturation process. One of the defining events of Stage VII is the release of mature spermatids from the seminiferous epithelium into the lumen of the tubules. This stage is marked by the presence of the most mature spermatids at the luminal edge of the epithelium. Furthermore, primary spermatocytes at the preleptotene/leptotene stage, which are just about to enter meiosis, are typically seen. Thus, we decided to choose these stages to avoid the high diversity of cell density derived from the difference in stages of spermatogenesis. We analyzed frozen testis sections stained with hematoxylin and eosin. The images were generated using nanoZoomer, and cells in stage VI-VII seminiferous tubules were counted. The number of cells localized in seminiferous tubules were divided by the area. We counted cells in a minimum of 6 tubules from 4 different control and 4 nicotine-exposed testis. We found that the cell density decreases by 0.7-fold (p = 0.03, nonparametric Mann-Whitney test) as a result of nicotine exposure (Fig. 1B).
Fig. 1
Testis morphology in Sprague-Dawley male rats prenatally exposed to nicotine. A Representative images of testes from mice exposed in utero to 0 (top) and 6 mg/kg/day (bottom) of nicotine. Sections were stained with H&E, scale bars are 100 µM (left panels) and 50 µM (right panels). B Number of cells per tubule. C Thickness of seminiferous tubules, n = 4 dose 0 mg/kg/day and n = 4 dose 6 mg/kg/day; significance, exact p-value are indicated on the top of the graph, the p -values were determined by the Mann-Whitney nonparametric test. Rats were sacrificed at 11 weeks of age. The cells were counted in stage (VI-VII) tubules. All plots in the Figure represent averaged values ± standard deviation
We also examined the thickness of the cell layer. We used the Fiji tool [21] and measured the parameter “perimeter”. We drew a line from the outer membrane of the seminiferous tubule to its center and measured the distance to the lumen. We compared the average values from at least 20 tubules from 4 different controls and 4 nicotine-exposed testes. We found a 0.78-fold (p = 0.03, nonparametric test) decrease in seminiferous tubule thickness at stage VI-VII in exposed animals compared to controls (Fig. 1C).
Next, we analyzed the number of germinal meiotic cells using the γH2AX marker. The non-canonical phosphorylated histone variant H2AX replaces the canonical histone H2A during formation of double-strand breaks (DSBs), thus allowing the chromosomes to become more condensed (reviewed in [22]). DSBs formation occurs throughout the whole genome and γH2AX marks are widely spread at each chromosome. At early stages of meiosis (leptotene-zygotene) when DSBs start to form, γH2AX shows a very bright staining all over the nucleus. At the later stage (pachytene-diplotene), when autosomes segregate and DSBs are getting repaired, γH2AX marks are retained only at sex-chromosomes as a bright spots [23]. γH2AX staining at cells such as Sertoli cells or type 2 spermatocytes, spermatogonia and spermatids is normally weak and generally only appears as foci corresponding to occurrence of random DNA damage. We performed γH2AX analysis on frozen testis sections. The sections were fixed with 4% PBS-buffered paraformaldehyde, washed and blocked, and the immunofluorescence assay was performed using an antibody against γH2AX (Fig. 2A, B) as described in the Methods section. The images were taken with objective 20 × and fixed time of exposure. We scored γH2AX positive cells (green staining) and compared them to the total number of cells in the tubules (DAPI staining). Our analysis revealed that nicotine exposure decreased the number of yH2AX-positive cells but the reduction was not statistically significant (0.8-fold decrease of γH2AX-positive cells, p = 0.06, nonparametric Mann-Whitney test, Fig. 2C). We also counted cells with a γH2AX pattern specific of leptotene-zygotene (bright staining all over the nucleus) and cells with a pattern specific of pachytene-diplotene (small bright spot in the nucleus). We found that nicotine exposure caused a non-significant decrease in the number of leptotene-zygotene-positive cells (p = 0.11, nonparametric Mann-Whitney test, Fig. 2D, left) and did not alter the number of pachytene-diplotene-positive cells (Fig. 2D, right). Thus, a small decrease in germ cell number, likely due to reductions in leptotene-zygotene and other cell types such as spermatogonia, could contribute to the decrease in total germ cell numbers even though the decrease in the number of leptotene-zygotene cells did not reach statistical significance.
Fig. 2
The effects of gestational nicotine exposure on meiotic and somatic testis cell. A Representative images of 11 week-old male testis spreads immunostained for γH2AX (green) in control (top) and nicotine (bottom) exposed testis; scale bar is 50 µM. B High magnification image (63X objective), z-leptotene-zygotene cells, p-pachytene-diplotene cells, scale bar is 20 µM. C Quantitative analysis of the ratio of γH2AX-positive to total number of cells; n = 4, dose 0, n = 5 dose 6 6 mg/kg/day, **p < 0.01 or p-value is indicated on the top of the graph, nonparametric Mann-Whitney test. D Quantitative analysis of leptotene -zygotene (left) or pachytene-diplotene (right) γH2AX-positive cells, p-value is indicated on the top of the graph, nonparametric Mann–Whitney test. E Analysis of gene expression in testis of 11 week-old rats by RT-qPCR; n = 5 dose 0, n = 6 dose 6 mg/kg/day. All plots represent average values ± standard deviation
To reveal which cell populations might be impacted by nicotine, we analyzed the expression of genes which are specifically expressed in each cell population based on a recently published dataset [24]. To this end, we extracted total RNA from whole testis and performed quantitative RT-qPCR analysis (Fig. 2E). Our analysis revealed that nicotine exposure decreased the expression of genes that are specifically expressed in spermatogonia, Kit and Lgr4, by 0.16- (p = 0.004), and 0.15-fold (p = 0.008), respectively. In contrast, the expression of Stra8 tended to increase 1.5-fold (p = 0.1). We also determined that nicotine exposure decreased the expression of several genes normally expressed in spermatocytes, Piwil1, Pttg1, H2ax and Rad51, by 0.15 (p = 0.004), 0.4 (p = 0.004), 0.1 (p = 0.004), and 0.27-folds (p = 0.004), respectively. The Kiss1 gene that is specific for Sertoli cells was increased by 2.13-fold (p = 0.004) by nicotine exposure; in contrast, Ctsl and Amhr2 genes which are also expressed in Sertoli cells were decreased by 0.6 (p = 0.008) and 0.08-folds (p = 0.004), respectively (Fig. 2E). Finally, Prm1, specific for the spermatid fraction was decreased by 0.48 (p = 0.004) by nicotine exposure (Fig. 2E). The most dramatic changes in gene expression were in the spermatogonia and spermatocyte fractions. Considering the relatively small decrease in germ cell numbers, we believe that these changes in gene expression reflect a decrease in these cell types.
In conclusion, the morphology analysis demonstrated that nicotine exposure causes a small but significant decrease in the germ cell population, with a tendency toward decrease of the leptotene-zygotene cells. We also observed perturbations in the expression of genes specific for each cell type, suggesting that prenatal exposure to nicotine alters gene expression in almost all cell types of the testis. However, the changes in expression of spermatogonia and spermatocytes genes could be due to a decrease in these cell populations.
Nicotine exposure perturbs meiotic progressionTo further explore effects of nicotine exposure on meiosis we analyzed the formation of H3K9me3 marks in nuclei. During meiosis H3K9me3 marks are normally localized all over the nucleus in the leptotene-zygotene stages and become concentrated at the nuclear periphery at the pachytene-diplotene stages [25]. H3K9me3 is normally enriched at regions of compact heterochromatin during meiosis (reviewed in [26]). Normal meiosis is characterized by patches of H3K9me3 marks mainly at the nuclear periphery, and these can be perturbed in certain pathological conditions in which alteration of telomere attachment and reduction of H3K9me3, leads to a loss of chromatin repression at telomeres [27]. To analyze H3K9me3 marks, cells were dissociated from whole testis and passed through a cell strainer to make cell spreads on slides. The spreads were fixed with paraformaldehyde, washed, blocked and immunostained for H3K9me3 (Fig. 3A) and SYCP3, a chromosome marker. Z-stack images were taken with similar exposure time and a quantitative analysis of the mark’s intensity was performed using imageJ as described in Methods. H3K9me3 and DAPI intensity were measured, and the H3K9me3 signal was normalized to DAPI staining. The analysis was performed for a minimum of 10 cells for each biological replicate (4 controls and 4 nicotine exposed), the averaged values were compared and plotted and the nonparametric Mann-Whitney test was used. We found a 1.7-fold increase in H3K9me3 staining in PNE cells, suggesting a possible impact of nicotine on normal chromosome segregation (Fig. 3B).
Fig. 3
The effect of gestational nicotine exposure on H3K9me3 levels and GRRs gene expression. A Representative images of 11 week-old male testis spreads immunostained for H3K9me3 (green) or SYCP3 (red, to visualize chromosome) in control (top) and nicotine-exposed mice; scale bar is 5 µM. B Quantitative analysis of H3K9me3 staining intensity; n = 5 dose 0, n = 6 dose 6 mg/kg/day, **p < 0.01, nonparametric Mann-Whitney test. C Quantitative analysis of GRRs gene expression in testis of 11 week-old males, n = 6 dose 0, n = 5 dose 6 mg/kg/day, **p < 0.01 or p-value indicated on the top of column, nonparametric Mann-Whitney test. All plots represent average values ± standard deviation
Next, we sought to analyze the expression of meiotic GRR genes given their importance for optimal meiosis. Our analysis showed that most of the tested GRR genes were increased by nicotine exposure: Brdt, Dazl, Ddx4, Tdrd1 displayed a 1.5-, 1.6-, 1.7-, and 5.0-fold increase, respectively, suggesting a potential perturbation of reprograming at GRR loci by nicotine exposure (Fig. 3C). Hormad1 expression which is essential for meiosis [28] also showed a trend towards increase by 1.2-fold (Fig. 3C).
In summary, meiosis and GRRs gene expression were perturbed in PNE male rats suggesting that PNE has a deleterious effect on the reproductive system in adults.
The H3K9me3 regulatory histone marks are increased in nicotine exposed testisSince nicotine exposure leads to changes in microbiota and to an increase in short acid chain groups known to increase acetylation of histones [12], we decided to analyze major regulatory histone levels which are important during meiosis [29]. The changes in histone acetylation could perturb other histone marks as histone acetylation and histone methylation often compete for same lysine residues. For instance, acetylation and methylation of a given lysine site are mutually exclusive, thus the methylation at that site can prevent activation by acetylation [30]. To check the level of modified histones in testis more precisely, we purified the histones from whole testis as described in the Methods section and performed the quantitative analysis of modified histones (Additional file 3: Fig S1). The signal of histone marks was normalized to Ponceau red staining (Additional file 3: Fig S2). First, we analyzed presence of acetylated histone H4. We did not observe a significant change in H4Ac levels in nicotine-exposed testis (Fig. 4A). We acknowledge here that the analysis was performed on adult testis while the exposure occurred during the embryonic development, hence we may be observing compensatory effects.
Fig. 4
Analysis of histone marks and expression of the spermatid marker ASB17 in testis of control and nicotine-exposed rats. A Representative images of H4Ac, B H3K4me3, C H3K9me3, and D ASB17. c1-c5 are controls and n1-n5 are nicotine-exposed samples, *p < 0.05 nonparametric Mann-Whitney test
Next, we analyzed the presence of H3K4me3 (which marks open chromatin). Our analysis showed no significant changes in the global level of H3K4me3 in response to nicotine exposure, although we cannot exclude the possibility of regional changes (Fig. 4B).
We also analyzed H3K9me3 mark which are associated with gene silencing and are abundant at compact heterochromatin. Here, in contrast, H3K9me3 marks were significantly increased in nicotine-exposed samples (Fig. 4C). The results of H3K9me3 marks detected by WB are consistent with the immunofluorescence results (Fig. 3A, B), which also demonstrated higher level of H3K9me3 in spermatocytes suggesting a global impact of nicotine exposure on H3K9me3.
To determine the effect of nicotine on haploid cell populations we analyzed the level of the ASB17 protein by WB (Fig. 4D). Asb17 is expressed exclusively in the testis, and Asb17 deficiency leads to a significant reduction of apoptosis in germ cells [31]. The highest expression of Asb17 is detected in round spermatids [32], suggesting its role in spermiogenesis. To analyze its expression, we extracted proteins from the testis and performed analysis by WB. We found that ASB17 levels were not significantly changed in nicotine-exposed cells (Fig. 4D), suggesting that the number of haploid cells is not notably perturbed, and changes in cell population are due to alterations of less mature cell types such as spermatocytes. We also performed the analysis of PRM2 levels. We found that PRM2 levels were not significantly changed in nicotine-exposed cells (Additional file 3: Fig S3).
In summary, higher level of H3K9me3 during meiosis caused by nicotine exposure can affect compact heterochromatin during meiosis. WB analyses suggest the absence of global changes in post-meiotic haploid cells, indicative of no developmental delay occurring post-meiosis.
Nicotine affects DNA methylation in the peripheral nervous system signaling and transcription factor genesTo explore the impact of nicotine on DNA methylation we performed DNA methylation analysis using a genome-wide sequencing technique. Briefly, we extracted DNA form testis and carried out DNA methylation enrichment experiments using the EpiMark enrichment kit (NEB). The kit is based on the purification of methylated DNA fragments bound to the methyl-CpG binding domain of human MBD2 protein, which is fused to the Fc tail of human IgG1, and coupled to protein A beads. After several wash steps, the enriched DNA sample is eluted from the beads and used for library construction. The library was built by using NEBNext® Ultra DNA Library Prep Kit (Illumina) using dual labeled multiplexed primers. The sequencing was performed by the Genomics platform, and obtained reads were filtered and aligned to the rat reference rn7 genome. The number of reads was normalized by size factors. The regions with peaks were determined using both methylated enriched and input libraries. The reads were counted at the peaks for each biological replicate and statistical test was performed. Nearly ~ 57,000 of methylated regions were detected. We identified 366 regions that showed differential pattern of DNA methylation (FDR < 0.1) in response to nicotine exposure (Fig. 5A, Additional file 1). Among differential peaks, 198 showed increased methylation and 165 regions showed decreased DNA methylation (Fig. 5B, Additional file 3). Analysis using the ChIPseeker package showed that most of the differentially methylated regions (DMRs) are genomic elements located distally from the gene promoters. However, nearly ~ 14% of DMRs were found in introns (Fig. 5C). To determine differentially methylated genes based on the DMRs, we assigned DMRs to genes using ChIPseeker. Functional annotations of DMRs by DAVID showed that they are enriched in genes that are expressed in neuronal cell body (Dmd, Tgfb1) (Fig. 5D, E); genes encoding transcription factors (Foxj3, Nfact2, Rfx3, Meis3, Rbpj, Foxs1, Dmtf1, Hoxb3, Hoxa2, Tead1, E2f7, Hoxa5, Hoxb5, Creb5), the glutamatergic synapse proteins (Qsec2, Dtnbp1, Cabp1, Begain, Dnm2, Olfm1, Actc1, Slitrk2, Psd2, Prkar2a, Slitrk1, Syt10, Gpc6, Adgrl3), and GABAergic signaling factors (Slitrk2, Slitrk1, Sst, Iqsec3, Igsf9b) (Fig. 5F). In addition, the genes encoding estrous cycle-related factors (Trpm2, Anxa1, Cyp1b1, Pgr) harbored DMRs (Fig. 5F).
Fig. 5
Genome-wide DNA methylation analysis of testis from 11-week-old rats. A Heatmap of all annotated DMRs. Counts of genes with FC > 1.5 and FDR < 0.1 were log transformed and plotted in R using Pheatmap, and as input log2 counts of DMRs regions were used. B Volcano plot of DMRs. C Genomic localization of DMRs determined by ChIPseeker. The differentially methylated peaks within Tgfb1 (D) and Dmd (E) are shown in red dashed boxes. Plots from control (blue) and nicotine-exposed (red) samples are shown. Each control and treatment group contained three replicates. The sequencing reads were mapped to the reference rn7 Rattus norvegicus genome, normalized and converted to bedgraph files, which were visualized in IGV. The signal intensity is shown in brackets. F Functional annotation as “biological process”, “cellular localization” or “molecular function’ of genes located in common DMRs by DAVID. Bars were sorted by adjusted p-values and the length of each bar represents the number of genes in this group. G Normalized DNA methylation at the vicinity of Pgr gene, counts were extracted from sequencing data and plotted in Excel, exact p-value are indicated on top of columns. H Methylation -specific PCR. DNA from testis was bisulphite-converted and used for qPCR using primers specific for methylated and unmethylated DNA. ΔΔCq Method was used for the analysis of the ratio of methylated-to-unmethylated DNA. The average ratios were plotted, *p < 0.05. **p < 0.01, or exact p-value is indicated on top of the column, statistical significance was estimated by the nonparametric Mann-Whitney test
Notably, nicotine exposure caused a decrease in DNA methylation of the progesterone receptor gene (Fig. 5G). Testicular progesterone is a by-product of steroidogenesis which is not converted into testosterone. Serum 17-OHP appears to be a reliable proxy marker for intratesticular testosterone levels and could potentially be used as a readout to titrate or change medications that alter intratesticular testosterone. Based on this data we believe that nicotine exposure likely perturbs testosterone levels [33].
To confirm the DNA methylation state of GRRs by methylation-specific PCR, we performed bisulfite conversion of testis DNA and designed methylation-specific PCR primers. We used Methprimer (http://www.urogene.org/methprimer/index.html [34]) to design primers for methylated and unmethylated bisulfite-converted DNA PCR and checked the specificity of PCR products (Additional file 3: Fig S4A–D) by analyzing the melting curves. We compared the methylated-to-unmethylated ratios using the ΔΔCq method, and statistical significance of methylated-to-unmethylated PCR ratio was determined using the nonparametric Mann–Whitney test. This analysis confirmed that GRRs in nicotine-exposed mice have a lower DNA methylation level (e.g., Dazl, Hsf5, Phdlc1, Brdt) (Fig. 5H). These changes negatively correlate with gene expression, suggesting the strong impact of nicotine on GRRs.
Due to their importance for meiosis, we specifically examined the DNA methylation status of 45 GRRs. We extracted the sequencing read numbers of the methylated regions of GRRs and plotted them in Excel (Fig. 6A, Additional file 3:S1). We found that DNA methylation marks are present at ~ 30 genes. We observed that DNA methylation has a tendency to decrease at the Hsf5 (p = 0.1, nonparametric test) and is increased at the Mov10l1 gene (p = 0.1, nonparametric test) in response to nicotine exposure (Fig. 6B). Notably, decreased DNA methylation at GRRs negatively correlates with GRRs gene expression (Fig. 3C) suggesting a possible impact of PNE on DNA methylation at GRRs during development and on expression of these genes in adulthood.
Fig. 6
DNA methylation analysis of GRRs. A Normalized DNA methylation of GRRs. Counts were extracted from sequencing data and plotted in Excel, exact p-value are indicated on top of columns (nonparametric Mann–Whitney test, FDR test was not applied). The differentially methylated peaks near B Hsf5 and C Mov10l1 are shown in red dashed boxes. Plots from control samples are shown in blue, plots from nicotine-exposed samples are shown in red. Each control and treatment groups contained three replicates. The signal intensity is shown in brackets. D Motif analysis by MEME-ChIP revealed two enriched motifs, parts of these motifs are significantly similar to NRF1 and ETV4 binding sites
We also searched for the presence of motif enrichments at DMRs to identify a possible mechanism of action of PNE. To this end, we extracted sequence information of DMRs in fasta format and performed a motif search using MEME-ChIP which performs comprehensive motif analyses (including motif discovery) on sequences in which motif sites tend to be centrally located, such as ChIP-seq peaks. We used the eukaryotic motif database MOUSE/HOCOMOCOv11 for the search. Our analysis determined that two common motifs are found in many sequences. Part of the first common motif is similar to the binding site for the NRF1 transcription factor (Fig. 6D). Nrf1 encodes a regulator of mitochondrial metabolism that plays a critical role in the development of post-migrating primordial germ cells (PGC) [35], suggesting the role of this factor in nicotine-treated germ cells. The conditional ablation of NRF1 in gonocytes dramatically down-regulates several germline genes (Dazl, Lin28a, Ddx4), blocks germ cell proliferation, and subsequently leads to male infertility in mice [36] suggesting an important role of NRF1 in regulation of some GRRs.
Part of the second identified motif is similar to the binding site for ETV4 (Fig. 6E). ETV4 regulates cell proliferation [37]. Etv4 mutant mice fail to form specific motor neurons, which do not branch normally within their target muscles, and the cell bodies of neurons are displaced within the spinal cord [38]. Thus, it is possible that the disfunction in ETV4 binding caused by nicotine could affect the formation and functioning of the peripheral nervous system in many organs including the testis. In this respect, while Etv4(–/–) males show normal mating behavior, they do not leave copulatory plugs and sperm is not detected in the uteri of females that mate with Etv4(–/–) males [39].
In summary, DNA methylation analysis uncovered that upon nicotine exposure there is differential methylation of genes relevant to nervous system signaling and transcription factor activity. We propose that abnormal activities of the NRF1 and ETV4 transcription factors could alter the development and proliferation of post-migratory primordial germ cells.
RNA-seq analysis of the pituitary gland from nicotine-exposed animals reveals effects on genes important for cell migration, cell adhesion and GABAergic signaling.To explore the molecular mechanisms of nicotine exposure in the neuroendocrine system, we performed transcriptomic analysis of the pituitary gland using paired-end stranded RNA sequencing using 3 biological replicates from the nicotine-treated and control rats. We chose to study the pituitary gland due to its key role for hormonal regulation of reproductive functions. Total RNA was extracted, treated with DNAse I, and strand-specific libraries were prepared and sequenced in multiplexed mode. Reads were mapped to the reference rn7 genome. The differentially expressed genes were determined (Additional file 2). The samples showed some variations between replicates (Additional file 3: Fig S5A, B), possibly due to the complexity of cellular milieu in the pituitary. However, we were able to identify 60 differentially expressed genes (DEGs) between nicotine exposed samples and controls (FDR < 0.1) (Fig. 7A, Additional file 3: Fig S5C, Additional file 3: Table S1). The majority of DEGs were downregulated in nicotine treatment samples (Fig. 7A, Additional file 3: Fig S5D). We performed a functional annotation of the DEGs with the gene ontology program DAVID. The strongest enrichment in DEGs was found in genes related to Tgfb1 signaling, regulation of cell migration, and cell adhesion among others (Fig. 7B). It is noteworthy, that Tgfb1 regulated genes (Acta2, Col4a2) were upregulated by nicotine exposure in the pituitary gland, in contrast to the decrease in Tfgb1 DNA methylation observed in the testis of exposed animal, suggesting a possible link between pituitary and testis Tfgb1 signaling pathways.
Fig. 7
RNA-seq analysis in the pituitary gland. A Heatmap of all annotated DMRs. Normalised transcript per million counts (TPM) of genes (FDR < 0.1) were log transformed and plotted in R, c1-c3 control, n1-n3 nicotine exposed samples, upregulated genes are in red, downregulated genes in blue. B Functional annotation “biological process”, “cellular component” or “molecular function” of DEG genes according to DAVID, bars sorted by p-values, and each bar represents the number of genes in each group, the analysis was done without multiple testing correction. C Heatmap of GABAergic signalling genes. The TPMs were log transformed and plotted in R. D The network of GABAergic signalling genes, red line—indicates the presence of fusion evidence, green line—neighbourhood evidence, blue line—cooccurrence evidence, purple line—experimental evidence, black line—coexpression evidence, network was built by String (https://string-db.org/cgi/). E Methylation-specific PCR in the pituitary gland. DNA from pituitary glands was bisulphite-converted and used for qPCR using primers specific for methylated and unmethylated DNA. The ΔΔCq method was used for analysis of the ratio of methylated-to-unmethylated DNA. The averaged ratios were plotted, *p < 0.05, statistical significance was estimated by nonparametric Mann-Whitney test
Since we observed some alteration in DNA methylation of GABAergic signaling genes in the testis, we decided to take a closer look at these genes in the pituitary gland RNA-seq data. To this end, we catalogued genes that are combined by the gene ontology term “GABAergic synapse” using the AMIGO database (https://amigo.geneontology.org/amigo). 136 genes were found related to this term, with 121 out of them expressed in the rat pituitary gland. We determined that Erbb4 and Slc6a6 were significantly upregulated upon nicotine exposure and three other genes (Slc6a11, Slc32a1, Slc6a6) showed a tendency to increase. In contrast, six other genes (Gabra4, Gabrg2, Epha3, Gabra3, Gap43, Adra2a) showed a tendency to decrease (Fig. 7C). These altered genes encode for proteins that are combined in network with central players GABRA2, GABRA3, GABRA4 and GABRG2 (Fig. 7D).
Similar to our approach with testis GRRs genes’ analysis, we decided to explore DNA methylation in some DEGs using methylation-specific PCR. We extracted pituitary gland DNA and performed the bisulfite conversion. Primers for methylation specific PCR were designed and qPCR analysis was carried out. Our results demonstrated that nicotine exposure is associated with increased DNA methylation in intron 1 of Erbb4 gene (Fig. 7E), within the CpG region located in the gene body. Importantly, methylated CpG islands in the gene body positively correlate with gene expression [40]. We also found an increase of DNA methylation in the promotors of Gjb2 and Efemp1 genes which negatively correlates with gene expression (Fig. 7E). We chose to analyze these genes due to their importance for the brain function. Mutations in Gjb2 gene are responsible for the recessive deafness [41, 42]. Moreover, both nicotine and smoke exposure are strongly associated with the risks for developing sensorineural hearing loss [43]. Study showed that EFEMP1 is a paracrine activator of Notch signaling in endothelial cells and promotes glioma angiogenesis [44]. The increase in DNA methylation of these genes is negatively correlate with the gene expression, suggesting that that nicotine exposure during embryonic development could perturb the epigenetic regulation of these genes.
In summary, RNA-seq analysis of the pituitary showed that expression of genes related to Tgfb1 signaling, cell migration, cell adhesion and GABAergic signaling was altered between the control and the nicotine-exposed group, suggesting that PNE affects the neuroendocrine system as well. DNA methylation of several DEGs was similarly altered suggesting that gestational exposure to nicotine could also induces changes in the pattern of DNA methylation in the pituitary.
留言 (0)