
記住我



The kidney requires a high density of functional mitochondria to maintain its normal function. It is known that mitochondrial dysfunction is linked to chronic kidney disease (CKD), acute kidney injury(AKI), and other kidney diseases.[1] Indeed, mitochondrial metabolic disorders in glomerular podocytes, mesangial cells, and endothelial cells can cause renal dysfunction.[2,3] The cellular stress response is associated with broad molecular changes that safeguard cells against short-term damage and enable them to survive in long-term adverse conditions.[4] Pathogens, tumorigenesis, and mechanical damage are distinct cellular stressors that can induce abnormal double-stranded DNA (dsDNA) localization within cells. Several compensatory mechanisms, such as repairing unstable genomes and degrading unfolded proteins, allow cells to respond appropriately to mitochondrial stress.[5] However, when these rescue measures fail, damaged mitochondria release mitochondrial DNA (mtDNA) into the cytoplasm, which is a significant source of damage-associated molecular patterns (DAMPs).
DAMPs are endogenous molecules released by damaged or dying cells, according to their original definition,[6] which in turn induce strong innate immune responses.[7] When sensing stress signals or responding to damage, mitochondria release their oxidized and cleaved mtDNA into the cytoplasm. Most organisms detect aberrant DNA via innate immune receptors. Toll-like receptor 9 (TLR9) as the first identified DNA sensor was discovered by Hemmi et al[8] in 2000. Since then, several cytosolic DNA sensors, including Z-DNA binding protein 1 (ZBP1), absent in melanoma 2 (AIM2), interferon-inducible protein 16 (IFI16), and the cyclic guanosine monophosphate-adenosine monophosphate (GMP-AMP) synthase (cGAS), have been identified.[9] Among the various DAMPs that can alert innate immune mechanisms to kidney damage, aberrant DNA recognition by the cGAS–stimulator of interferon gene (STING) signaling pathway is particularly crucial for the intrinsic sensing of cells.[10] In this review, we summarize the mechanisms underlying mtDNA leakage resulting from mitochondrial damage, with a specific focus on the correlation between mtDNA-mediated cGAS-STING signaling and the pathogenesis of kidney diseases. Furthermore, we present latest findings of cGAS-STING signaling pathway in cell, with a particular emphasis on its downstream signaling related to kidney diseases. Through this review, we hope to enhance our understanding of the intricate links among the cGAS-STING signal pathway, mitochondrial damage, and kidney diseases, and provide new perspectives and approaches for the prevention and treatment of kidney disease.
Immunogenicity of MtDNAUnder normal physiological conditions, mtDNA is located adjacent to the inner mitochondrial membrane (IMM),[11] where distinct DNA repair mechanisms maintain genetic integrity by selectively amplifying intact copies of mtDNA.[12] Mitochondria are believed to have originated from eubacteria; thus, mtDNA is often unmethylated and differs from nuclear DNA.[13] Moreover, mitochondria lack the sophisticated mechanisms used to repair genomic DNA, making mtDNA more vulnerable to the effects of reactive oxygen species (ROS) and environmental toxins.[14] Environmental, genetic, and spontaneous damages, such as ionizing/non-ionizing radiation, alkylating agents, ROS, lipid peroxides, endogenous chemicals, and the chemical instability of DNA, constantly threaten the integrity of nuclear DNA and mtDNA.[15] Mitochondria respond to cellular stress and homeostatic imbalance by releasing DAMPs, including oxidized mtDNA, ROS, N-formyl peptide, and cardiolipin.[16] The recognition of aberrant DNA, particularly ectopic mtDNA, by the cGAS-STING signaling pathway plays a vital role in the innate immune response to kidney damage.[17–21]
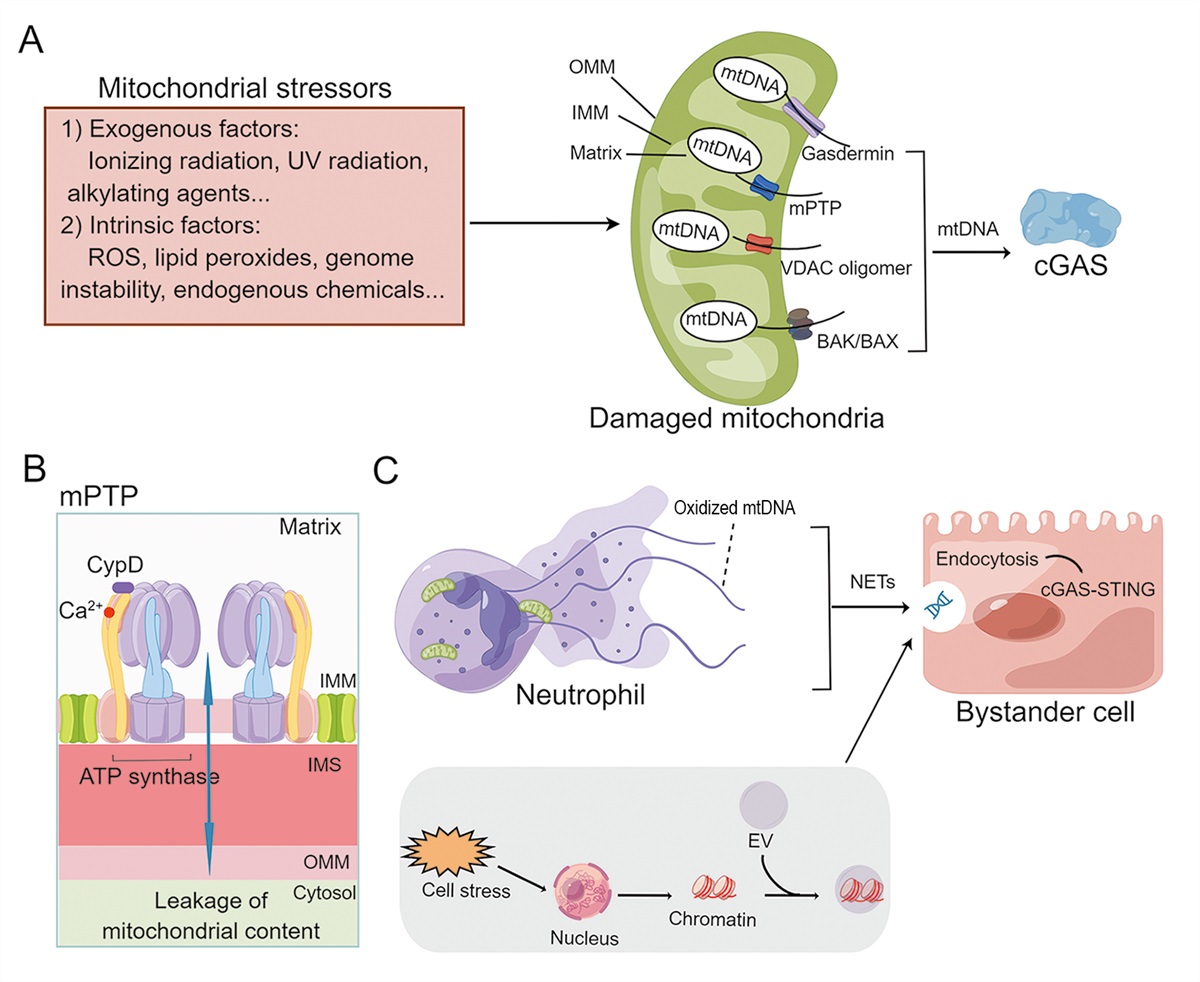
MtDNA Leakage Mechanism of mtDNA leakage into cytoplasmThe mtDNA leakage is intrinsically linked to mitochondrial dynamics, as well as changes in mitophagy and apoptosis, all of which can be influenced by different levels of stress within the cell.[22] The primary factor leading to the release of mitochondrial components into the cytoplasm is the loss of mitochondrial membrane integrity.[23] Four major pore-forming complexes are thought to facilitate the release of mtDNA from damaged mitochondria, as described in the sections below [Figure 1].
 Figure 1:
Figure 1: Mechanisms of mtDNA leakage. (A) Mechanisms of mtDNA leakage to the cytoplasm. Environmental and intrinsic genotoxic damage factors, including UV radiation, ionizing radiation, and alkylating agents, as well as ROS, lipid peroxides, endogenous chemicals, and genome instability, constantly threaten the integrity of the nucleus and mtDNA. As a response to cellular stress and homeostatic imbalance, mitochondria release DAMPs, including oxidized mtDNA. There are four major pore-forming complexes thought to facilitate the release of mtDNA from damaged mitochondria: VDAC pore, mPTP, BAK/BAX pore, and gasdermin pore. (B) ATP synthase-mediated mPTP opening. (C) Mechanisms of mtDNA leakage into the extracellular space. As a result of cell death and stress, mtDNA is released into the extracellular space via EVs or extracellular traps such as NETs. Subsequently, mtDNA is endocytosed by bystander cells and activates the cGAS-STING pathway to participate in intercellular interactions. ATP: Adenosine triphosphate; BAK: BCL2 antagonist/killer 1; BAX: BCL2 associated X; BCL2: B-cell lymphoma 2; cGAS-STING: Cyclic GMP-AMP synthetase stimulator of interferon genes; CypD: Cyclophilin D; DAMPs: Damage-associated molecular patterns; EVs: Extracellular vesicles; IMM: Inner mitochondrial membrane; IMS: intermembrane space; mPTP: Mitochondrial permeability transition pore; mtDNA: Mitochondrial DNA; NETs: Neutrophil extracellular traps; OMM: Outer mitochondrial membrane; ROS: Reactive oxygen species; UV: Ultraviolet; VDAC: Voltage-dependent anion channel.
Mitochondrial permeability transition pore and voltage-dependent anion channel (VDAC) oligomer poreThe mitochondrial permeability transition pore (mPTP) is a supramolecular structure located at the interface of the inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM), serving as a voltage- and Ca2+-sensitive channel.[24] Upon cells exposure to radiation, oxidative stress, or lipopolysaccharides (LPS), mitochondria release mtDNA fragments into the cytoplasm through the opening of mPTP.[25–28] Recent evidence emphasizes the significant role of mitochondrial F1Fo ATP synthase in the formation of high-conductance mPTP [Figure 1B].[29] The VDAC is one of the most abundant proteins in the OMM, which has two conductive states, i.e., an open, anion-selective state, and a closed, microcation-selective state, and play critical roles in mitochondrial signaling and calcium homeostasis.[30] Recent studies have shown that oxidatively stressed mitochondria release short fragments of mtDNA through pores formed by VDAC1 in a mouse model of systemic lupus erythematosus (SLE).[31] Furthermore,VDAC may regulate mPTP formation or opening in response to mitochondrial stress.[32]
BAK/BAX poreB-cell lymphoma 2 (Bcl-2)-associated X protein (BAX) and BCL2 antagonist/killer 1 (BAK), which are members of the Bcl-2 family, have been identified as key participants in the process of programmed cell death.[33] Studies have shown that the formation of pores in the OMM mediated by BAK and BAX is implicated in both pyroptosis and intrinsic apoptosis, resulting in the release of mitochondrial components into the cytosol.[34,35]
Gasdermin (GSDM) poreGSDM proteins have been identified as a key executioner of pyroptosis.[36,37] Gasdermin D (GSDMD) forms pores in the plasma membrane, which leads to cytokine release from cells and inflammatory cell death.[38] However, GSDMD is also highly expressed in the mitochondrial membrane, where it induces the release of ROS and causes mitochondrial damage.[39] Recent research has revealed that the internalized bacterial endotoxin LPS activates GSDMD, which subsequently leads to the formation of mitochondrial pores and the release of mtDNA into the cytoplasm of endothelial cells. This process, in turn, triggers the activation of the cGAS/STING pathway, which plays a pivotal role in modulating cellular responses to inflammation.[40]
Mechanisms of mtDNA leakage to extracellular spaceAs a result of cell stress and death, mtDNA is released into the extracellular space through two mechanisms: (1) the release of extracellular traps, such as neutrophil extracellular traps (NETs), which are composed of extracellular polymers produced by neutrophils; and (2) the formation of extracellular vesicles(EVs), which result from the fusion of mitochondria with vesicular structures, similar to those used in intracellular secretion. Subsequent endocytosis of extracellular mtDNA by bystander cells serves as a mode of horizontal information exchange, activating the cGAS-STING pathway.[41–43] This pathway can activate a host of inflammatory factors and immune cells, thereby altering the immune status of neighboring tissues [Figure 1C].
Overview of the cGAS-STING Signaling PathwaycGAS serves as a primary cytosolic DNA sensor, interacting with the adaptor STING, which is anchored as a transmembrane protein on the endoplasmic reticulum (ER) membrane.[9] On activation of the canonical cGAS-STING pathway by cytoplasmic dsDNA, the cGAS enzyme synthesizes cyclic dinucleotide (CDN) 2,3-cyclic GMP-AMP (cGAMP), which functions as a second messenger and binds to its receptor STING. Upon activation, STING oligomerizes and translocates from the ER to the Golgi apparatus and recruits TANK-binding kinase 1 (TBK1), leading to phosphorylation of interferon regulatory factor 3 (IRF3). Phosphorylated IRF3 forms dimers and enters the nucleus, resulting in transcription of IRF3 target genes and production of proinflammatory factors such as type 1 interferons. The oligomerization of STING also facilitates the activation of IκB kinase (IKK)/nuclear factor kappa B (NF-κB) and subsequently induces the expression of proinflammatory cytokines [Figure 2].[44] Once STING fulfills its function, it is transported to the lysosome where it is degraded to attenuate pathway activation.[45]
 Figure 2:
Figure 2: Overview of the cGAS-STING signaling. Various cellular stresses, such as pathogen infection, tumor development, and mechanical damage, can result in the mislocalization of dsDNA. Under these circumstances, the mtDNA, a significant endogenous DNA molecule, has the potential to be released into the cytoplasm. As a result of cytoplasmic dsDNA activation, cGAS synthesizes cGAMP to bind with receptor STING. Activated STING oligomerizes and translocates from the ER to the Golgi apparatus and recruits TBK1, leading to phosphorylation of IRF3 and IKK. Together, these cascade reactions initiate the transcription of proinflammatory cytokines, including type I IFNs.The oligomerization of STING also facilitates the activation of IKK/NF-κB, thereby inducing the expression of proinflammatory cytokines. ATP: Adenosine triphosphate; CDN: Cyclic dinucleotide; cGAMP: Cyclic GMP-AMP; cGAS: Cyclic GMP-AMP synthase; dsDNA: Double-stranded DNA; ER: Endoplasmic reticulum; GTP: Guanosine triphosphate; IFN: Interferon; IKK: IκB kinase; IRF3: IFN regulatory factor 3; mtDNA: Mitochondrial DNA; NF-κB: Nuclear factor kappa B; STING: Stimulator of interferon genes; TBK1: TANK-binding kinase 1.
Activation and Regulation of the cGAS-STING Signaling PathwayInitial models assumed that co-localization of cGAS and DNA in the cytosol defines the specificity of the pathway for recognizing non-self DNA. However, recent research has demonstrated that cGAS is also found in all cellular compartments (membrane, cytosol, and nucleus),[46–48] and this subcellular compartmentalization has been associated with the signaling specificity of cGAS.[49] The precise mechanism by which cGAS dissociates from subcellular structures and senses ds-DNA, including mtDNA, is not yet clear. cGAS recognizes dsDNA regardless of its sequence and is capable of recognizing various types of dsDNA, including both foreign and self-DNA. Recent studies have shown that human cGAS exhibits a preference for longer dsDNA molecules, at least 45 base pairs or longer, for optimal activation.[50] Upon binding to dsDNA, cGAS undergoes dimerization, forming a complex with two dsDNA molecules and two cGAS molecules. This structural rearrangement allows cGAS to catalyze adenosine triphosphate (ATP) and guanosine triphosphate (GTP), resulting in the generation of cGAMP as a second messenger [Figure 2]. Notably, the cGAS product in metazoans forms a unique covalent linkage between the γ-phosphate of ATP and the 2′-hydroxyl group of GTP, creating a 2′,5′-phosphodiester bond. This bond is more effective in activating STING signaling compared to the canonical 3′,5′-phosphodiester bonds found in bacterial cyclic dinucleotides (CDNs).[51] Zn2+ has been found to facilitate cGAS activation by promoting its phase transition in the presence of cytosolic DNA,[52] while Mn2+ enhances dsDNA recognition by cGAS and promotes the recognition of cGAMP and other bacterial CDNs by STING, which is crucial for cGAS-STING activation in vivo.[53] Two substitutions in the cGAS DNA-binding surface, hcGAS Lysine187 and Leucine195, are necessary for enhancing the ability of cGAS to discriminate short and long DNAs.[50] Additionally, it has been demonstrated that interferon regulatory factor 8 (IRF8) plays a crucial role in promoting effective activation of STING-mediated innate immune responses in monocytes, independent of its transcriptional function in monocyte differentiation.[54]
To prevent cGAS-mediated autoreactivity, it is crucial to continuously clear self-DNA originating from both intracellular and extracellular sources. The clearance is accomplished through the action of several factors including cytosolic 3′ repair exonuclease 1 (TREX1), lysosomal DNase II, and adenosine deaminase 2 (ADA2).[55] In addition, the recognition of intact genomic DNA is inhibited by nucleosomes and chromatin architectural proteins like barrier-to-autointegration factor 1, which competitively bind to nucleosome-free DNA and suppress cGAS activity.[55] Mitochondrial DNA, lacking unique chromatin features, is physically separated to prevent its sensing.[56] ATP-binding cassette, sub-family C member 1(ABCC1) has been identified as a transporter that facilitates the ATP-dependent export of cGAMP, serving to modulate cellular STING responses and control cGAS-related autoimmunity in vivo.[57] The ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP1) is supposed to be the dominant 2′3′-cGAMP extracellular hydrolase, however, the mechanism by which cGAMP is transported to the extracellular space remains unknown.[58] When cGAMP binds with STING, STING undergoes a conformational change that facilitates the recruitment and activation of downstream signaling molecules, ultimately leading to the activation of STING-mediated immune responses. After STING accomplishes its signaling function, it is rapidly degraded, possibly through autophagy.[59] These safeguarding mechanisms ensure a balanced and transient innate immune response triggered by cGAS and STING. However, the malfunctioning of these safeguards can lead to constitutive or prolonged activation of innate immunity, contributing to acute and chronic kidney injury.[10]
Signaling Downstream of STING Activation Related to Kidney Diseases STING-PERK-eIF2α signalingA evolutionarily primitive STING-Protein kinase R-like endoplasmic reticulum kinase (PERK)-eukaryotic initiation factor 2α (eIF2α) signaling pathway has recently been discovered in the ER, which is completely independent of the traditional STING-TBK1-IRF3 signaling pathway.[60] Mechanistically, upon binding to cGAMP, STING associates with the ER membrane and interacts with PERK through its cytoplasmic carboxyl-terminal domain, leading to PERK activation. Notably, the STING-PERK-eIF2α signaling pathway is rapidly activated within the ER, occurring earlier than the classical STING-TBK1-IRF3 pathway, which takes place in the Golgi apparatus. Functionally, STING-mediated PERK activation leads to robust phosphorylation of eIF2α, selectively regulating the translation of messenger RNA (mRNA) and facilitating translation programs associated with inflammatory and survival signaling [Figure 3A]. Moreover, further investigations have demonstrated that pharmacological interventions targeting this non-classical cGAS-STING pathway can effectively mitigate cellular senescence triggered by injury, and attenuate fibrotic processes in the lungs and kidneys. These findings provide valuable insights into the crucial role of the cGAS-STING-PERK-eIF2α signaling pathway in regulating cellular senescence and kidney fibrosis.[60]
 Figure 3:
Figure 3: Signaling downstream of STING activation related to kidney diseases. (A) After binding with cGAMP, STING undergoes a conformational alteration and associates with PERK, a protein kinase found in the ER. This interaction leads to robust activation of STING. Subsequently, PERK phosphorylates eIF2α, which initiates a selective mRNA translation program that plays a crucial role in cellular senescence and renal fibrosis. (B) The activation of STING leads to the initiation of the COPII complex assembly, resulting in the lipidation of LC3. This lipidation process is facilitated by the involvement of WIPI-2. Subsequently, the lipidized LC3 molecules encapsulate damaged DNA, bacteria, or viruses. In murine models of DKD and Alport syndrome, there is evidence suggesting that the activation of STING in podocytes is associated with increased apoptosis and autophagy. This is supported by elevated protein levels of LC3B and Beclin-1. (C) Upon activation, STING translocates from the ER to the Golgi apparatus and recruits TBK1, leading to phosphorylation of IRF3 and the activation of IKK/NF-κB and subsequently induces the expression of proinflammatory cytokines. cGAMP: Cyclic GMP-AMP; COPII complex: Coat protein complex II; DKD: Diabetic kidney disease; eIF2α: Eukaryotic initiation factor 2α; ER: Endoplasmic reticulum; ERGIC: ER-Golgi intermediate compartment; IRF3: IFN regulatory factor 3; IKK: IκB kinase; mtDNA: Mitochondrial DNA; mRNA: Messenger RNA; LC3: Microtubule-associated protein 1 light chain 3; NF-κB: Nuclear factor kappa B; PERK: Protein kinase R-like endoplasmic reticulum kinase; STING: Stimulator of interferon genes; TBK1: TANK-binding kinase 1; WIPI-2: WD repeat domain phosphoinositide interaction 2.
STING induces autophagy related signalingRecent research has discovered that the induction of autophagy through STING trafficking represents a primordial function of the cGAS pathway.[61] The induction of autophagy through the cGAS-STING signaling cascade can be elucidated as follows: The activation of cGAS is triggered by dsDNA originating from either internal or external sources, resulting in the production of cGAMP. Subsequently, cGAMP binds to STING, thereby initiating the assembly of a complex involving secretion-associated Ras-related GTPase 1 (SAR1) and SEC24 homolog C (SEC24C), commonly known as the coat protein complex II (COPII complex). The COPII complex undergoes translocation from the endoplasmic reticulum (ER) to the ER-Golgi intermediate compartment (ERGIC), where the process of lipidation of microtubule-associated protein 1 light chain 3 (LC3) occurs. This lipidation process is facilitated by the involvement of WD repeat domain, phosphoinositide interacting 2 (WIPI-2). Subsequently, the lipidated LC3 molecules encapsulate damaged DNA, bacteria, or viruses, leading to the formation of autophagosomes [Figure 3B]. Ultimately, these autophagosomes merge with lysosomes, where their contents are degraded by various enzymes. In murine models of diabetic kidney disease (DKD) and Alport syndrome, there is evidence indicating that the activation of STING in podocytes is linked to increased apoptosis and autophagy, as evidenced by elevated protein levels of LC3B and beclin 1.[62]
STING-TBK1/IKK-NF-κBResearch on mouse models of DKD and AKI has shown that mtDNA leakage mediated by BAX triggers the activation of the STING-TBK1/IKK-NF-κB pathway, which often leads to kidney cell damage and inflammation.[18,19] In the DKD mouse model, lipotoxicity-triggered mitochondrial damage in podocytes leads to the leakage of mtDNA into the cytosol via BAX.[19] Interestingly, this BAX-mediated cytosolic mtDNA leakage is found to activate the cGAS-STING pathway, thus inducing podocyte damage, even in the absence of lipotoxicity. Notably, this process is linked to the activation of STING downstream effectors TBK1 and NF-κB, but not IRF3 [Figure 3C].[19] Furthermore, in the cisplatin-induced AKI mouse model, it has been observed to induce mtDNA leakage into the cytosol of tubules, possibly via BAX pores in the mitochondrial outer membrane. This subsequently triggers the STING-TBK1/IKK-NF-κB signaling, leading to inflammation and the progression of AKI.[18] In mitochondrial transcription factor A (TFAM) knockout (KO) mice, mitochondrial damage leads to the escape of mtDNA into the cytosol, which activates the STING-TBK1/IKK-NF-κB sensing pathway, mediating renal inflammation and fibrosis.[17]
MtDNA-cGAS-STING Pathway in Kidney DiseasecGAS-STING is a crucial innate immune pathway that acts as an early warning system against viral infections or other cellular stressors by initiating an immune response against foreign DNA or self-DNA. However, dysregulation of this pathway following the abnormal accumulation of mtDNA, as a result of various genetic or environmental factors, has a detrimental effect on the body, leading to various diseases and complications, including AKI, Lupus nephritis (LN), and CKD. Thus, exploring the role of the mtDNA-cGAS-STING axis in the pathogenesis of kidney disease may lead to the development of novel treatment strategies.
MtDNA-cGAS-STING pathway in AKIAKI is a complex and rapidly progressive disorder often caused by damage to the renal parenchyma. It is believed that mitochondrial dysfunction is an early event in AKI and plays a key role in its pathogenesis.[63] A burst of ROS during reperfusion of ischemic tissues can trigger mPTP opening and, thus, theoretically, increased mtDNA release.[64] Recent studies have explored the mechanisms and consequences of mtDNA release in AKI. For instance, it has been shown that increased mtDNA levels in the plasma of patients with sepsis-induced AKI are a hallmark of renal mitochondrial injury.[65] Similarly, patients with severe sepsis-induced AKI have significantly elevated urine mtDNA levels, which are positively correlated with plasma creatinine, urine neutrophil gelatinase-associated lipid, and kidney injury molecule-1 levels.[66] Furthermore, a previous study found that the severity of AKI was reduced using cGMP-selective phosphodiesterase inhibitors.[67] Feng et al[68] reported that the levels of receptor-interacting protein kinase 3 (RIP3) in the cytosol were increased following ischemia-reperfusion (I/R) in mouse kidneys. Mechanistically, RIP3 translocates to the mitochondria, where it interacts with mitofilin (a mitochondrial inner membrane protein) and promotes its degradation, resulting in increased mitochondrial damage and mtDNA release into the cytosol in an mPTP-independent manner. As a result of mtDNA release, the cGAS-STING pathway is activated, leading to increased nuclear transcription of proinflammatory markers, which aggravate kidney I/R injury.[68] In an animal model of cisplatin-induced AKI, Maekawa et al[18] observed that cisplatin induced mtDNA leakage into the cytosol of tubule cells, leading to the activation of the cGAS-STING pathway, thereby triggering inflammation and AKI progression. Meanwhile, the cisplatin-induced inflammatory response was attenuated in STING-KO mice. In another animal study, H151, a small-molecule STING antagonist, alleviated cisplatin-induced kidney damage and inflammation.[69] Moreover, in human kidney cells (HKCs), triptolide (TPL) induced ROS production, leading to mtDNA leakage, while also activating cGAS-STING signaling in mouse kidney tubules; the use of STING inhibitors alleviated kidney damage in these TPL-treated mice.[70] The above studies illustrate that mitochondrial dysfunction and activation of the mtDNA-cGAS-STING pathway are major inducers of kidney damage.
MtDNA-cGAS-STING pathway in CKDThere appears to be a link between mtDNA-cGAS-STING and chronic inflammation.[71] During mitochondrial dysfunction, the communication between mitochondria and the ER is disrupted, resulting in kidney tubule inflammation and fibrosis; this causes the progression of AKI to CKD.[72] Compared with healthy kidneys, the kidneys of individuals with CKD have significantly defective mitochondrial dynamics[73] and high levels of mitochondrial oxidative stress.[74] In mouse models of renal fibrosis, mitochondrial loss and decreased ATP levels were observed in juvenile animals; however, renal fibrosis and azotemia did not develop until adulthood.[17] Urine mtDNA levels were inversely correlated with estimated glomerular filtration rate (eGFR) yet positively correlated with interstitial fibrosis[75] and may therefore serve as a surrogate marker for permanent kidney damage in patients with non-diabetic CKD.[76] In the TFAM-KO renal fibrosis mouse model, mtDNA leakage into the cytosol activated the DNA-sensing cGAS-STING pathway, resulting in cytokine expression and immune cell recruitment.[17] Consequently, the genetic depletion or inhibition of STING reduced the severity of kidney fibrosis in mouse models of CKD.[17] Zhang et al[60] identified the STING-PERK-eIF2α pathway as a critical and physiological factor contributing to the progression of renal fibrosis in a surgically-induced mouse model of unilateral ureteral obstruction.
Diabetes is a leading cause of kidney failure, and DKD (also known as diabetic nephropathy) is a major trigger for CKD. The progression of DKD is associated with excess ROS production, activation of apoptosis, and defects in hyperglycemia-induced mitochondrial autophagy. It is well known that mitochondria play a central role in the pathogenesis of DKD.[77–79] For instance, urinary mtDNA content is markedly elevated in both diabetic patients and mice. Moreover, the intravenous injection of mtDNA into mice induced inflammation and subsequent kidney injury in these animals.[80] Additionally, mitochondrial dysfunction is associated with the severity of DKD.[81] When mesangial cells are exposed to high glucose concentrations, an increase in intracellular mtDNA content, accumulation of ROS, and increased mitochondrial damage can be observed.[82] In mice with DKD, kidney podocytes experience mitochondrial dysfunction and damage, which results in the leakage of mtDNA into the cytosol through BAX-mediated macropores. Cytosolic mtDNA in turn activates the cGAS-STING/TBK1/p65 pathway, leading to the production of inflammatory cytokines and podocyte injury.[19] It has been reported that activating STING in wild-type mice leads to albuminuria and podocyte loss, while the cGAS-STING signaling pathway is upregulated in mice with experimental DKD and Alport syndrome.[62] Both genetic depletion and inhibition of STING inhibit the progression of glomerular diseases and preserve renal function, demonstrating that the cGAS-STING pathway plays a vital role in mediating glomerular dysfunction.[62] Inflammatory cytokine production and podocyte damage occur as a result of the cytosolic-mtDNA-mediated activation of the cGAS-STING-TBK1-p65 pathway; consequently, the inhibition of STING can reduce podocyte injury in diabetic mice.[19] It has been reported that the sacubitril/valsartan combination or valsartan alone inhibits auto-DNA-activated cGAS-STING signaling in mice with DKD.[83]
These findings provide valuable insights into the molecular mechanisms underlying the development and progression of CKD, and suggest that targeting the cGAS-STING pathway may have therapeutic potential in CKD.
MtDNA-cGAS-STING pathway in SLE and LNSLE is characterized by multi-organ inflammation and the presence of circulating antigen-autoantibody immune complexes. It was reported that mitochondrial genetic alterations are related to the occurrence of SLE,[84] and that novel mtDNA genetic variants are associated with SLE susceptibility and prognosis.[85] mtDNA deposits have been found in the NETs of LN kidney biopsy specimens.[86] Moreover, SLE patients have higher levels of anti-mtDNA antibodies than healthy individuals, which is associated with the disease activity index in patients with LN.[87] According to one study, ROS are an important inducer of oxidized mitochondrial DNA (Ox-mtDNA) production by neutrophils in response to ribonucleotide immune complex stimulation in SLE patients.[41] Simultaneously, the authors found that SLE pathogenesis was linked to the inadequate degradation of neutrophil Ox-mtDNA both in vitro and in vivo.[41] Importantly, a significant increase in the production of STING-pathway-associated inflammatory cytokines could be observed in cells stimulated with Ox-mtDNA.[41] Similarly, neutrophils in SLE have reduced levels of protein kinase A (PKA) activation and mitochondria transcription factor A (TFAM) degradation, leading to the expulsion of immunogenic Ox-mtDNA into the extracellular space.[43]
Recent studies have found a defect in the mechanism of programmed mitochondrial clearance in SLE, which results in the accumulation of mitochondria-positive (Mito+) red blood cells (RBCs) in SLE patients, a marker of disease activity.[88] By activating cGAS in macrophages, antibody-mediated internalization of Mito+ RBCs induces the production of type I IFNs.[88] In SLE monocytes, accumulated mtDNA escapes autophagy and acts as an autocytoplasmic DAMP. This results in the activation of STING, which leads to tumor necrosis factor and interleukin-6 secretion and the subsequent proliferation of autoreactive lymphocytes.[89] In addition, the cGAS-STING pathway is activated by apoptosis-derived membrane vesicles in the serum of patients with SLE.[90]
Overall, the above studies prove that Ox-mtDNA is not simply a disease biomarker but also contributes to the development of SLE and LN via the cGAS-STING pathway.
Therapeutic Targeting of the MtDNA-cGAS-STING Signaling Pathway in Kidney DiseaseThe role of the mtDNA-cGAS-STING pathway in kidney disease has sparked significant interest in developing drugs that target mtDNA leakage and cGAS-STING pathway proteins. Many potential drug candidates have been identified for mitigating palmitic acid (PA)-induced podocyte damage and improving kidney function, including low concentrations of ethidium bromide, which blocks the replication and transcription of mtDNA, and BAI1, an inhibitor of BAX, which blocks PA-induced cGAS-STING activation.[19] Cyclosporin A (CsA) is a commonly used immunosuppressant in kidney disease and an inhibitor of Cyclophilin D (CypD), which prevents mPTP opening. Bendavia inhibited mPTP opening in the porcine ather
留言 (0)