
記住我
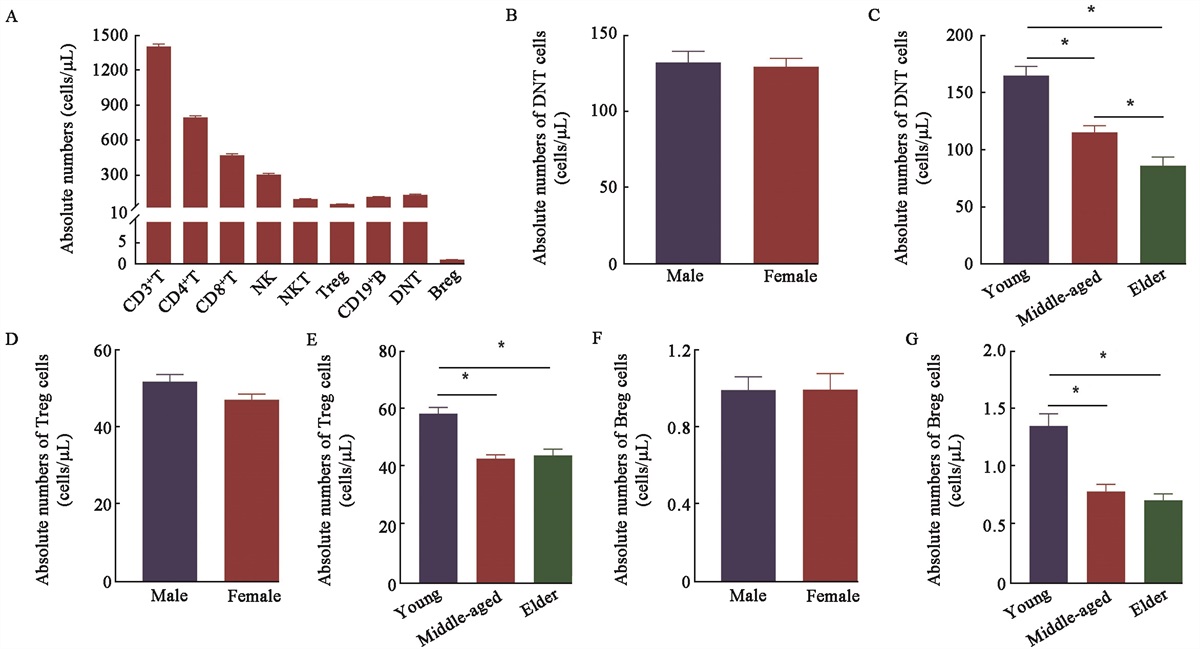








The incidence of lung cancer remains high, making it one of the most frequently diagnosed malignancies. Non-small cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer cases. Lung adenocarcinoma (LUAD) and squamous carcinoma of the lungs are the most prevalent subtypes of NSCLC.[1] Fifty-seven percent of patients with lung cancer are diagnosed with metastatic disease, and their 5-year survival rate is 6%.[2]
Liver is a common site of NSCLC metastasis, affecting approximately 20% of NSCLC patients.[3] Liver metastasis (LM) can modulate systemic immune responses,[4] leading to a poor prognosis. Patients with LM demonstrate a median overall survival (OS) of 3–4 months,[5] which is worse than that of patients with metastases in other organs, including the brain (7 months), bone (6 months), and lymph nodes.[6,7] For LMs associated with NSCLC, treatment includes surgical resection, transarterial chemoembolization, radiofrequency ablation, molecularly targeted agents (targeting the mutated epidermal growth factor receptor [EGFR], rearranged anaplastic lymphoma kinase [ALK], and rearranged reactive oxygen species [ROS] proto-oncogene 1, receptor tyrosine kinase), palliative radiotherapy, and immunotherapy.[8] Patients with LMs who undergo targeted or cytotoxic therapy[9] or surgery[10] have poor prognoses. Moreover, LMs negatively affect the responses to immunotherapy.[11] For instance, pembrolizumab,[12] nivolumab,[13] or durvalumab[7] is significantly less effective in NSCLC patients with LMs than in those without LMs.
Generally, immune responses to intrahepatic tumor antigens result in the systemic inhibition of antitumor immunity.[12] Cancer cells that enter the liver are exposed to a complex and distinct microenvironment (dendritic cells [DCs], Kupffer cells [KCs], and sinusoidal vessels with immunosuppressive functions) that may contribute to tumor evasion of immune surveillance during the course of immunotherapy. Hepatic parenchymal and non-parenchymal cells, together with recruited immune and inflammatory cells, are involved in the response to invasive tumor cells and may favor or suppress the progression of metastasis.[14] Compared with conventional antigen-presenting cells (APCs) in the steady-state liver environment, such as DCs, unconventional APC populations, constituting KCs, liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HepSCs), and hepatocytes, express low levels of major histocompatibility complex (MHC)-I/MHC-II and co-stimulatory molecules.[15] The liver has unique immunological properties, leading to a systemic loss of T cells, including T-cell anergy or apoptosis, regulatory T-cell (Treg) induction, or effector T-cell elimination mediated by conventional and unconventional APCs.[16]
The pivotal contribution of the liver microenvironment to the promotion of cancer cells and their growth within the liver suggests that targeting molecules and cells in the liver microenvironment is a preventive and therapeutic strategy for LM.[17] Targeting the factors of the tumor microenvironment (TME) alone or in combination with tumor-directed biotherapy or chemotherapy is advantageous based on the following reasons: (1) cancer cells rely on a supportive microenvironment for survival and growth, (2) the TME is composed of genetically stable cells that have more predictable responses and characteristics, and (3) targeting the TME may be advantageous for various cancer types, particularly for tumors that predominantly metastasize to the same location, for instance, the liver.[17]
This review provides insights into the epidemiology, physiology, pathology and immunology of LMs in patients with NSCLC. We focused on investigating the mechanisms by which certain etiologies alter hepatic homeostasis and addressed how such alterations facilitate tumor metastasis and modulate the surveillance of primary tumors or aggressive metastatic cells. We also explored the rationale and strategies for targeting the liver microenvironment to prevent and treat LMs.
Epidemiology and Prognosis of NSCLC-related LMsThe three most common organs involved in NSCLC metastasis are the adrenal glands, liver, and lungs.[4] LMs occur in approximately 4–20% of patients with NSCLC and 17% of patients with SCLC.[3,9,18] Data derived from the Surveillance, Epidemiology, and End Results database indicate that 5.1% of patients present with synchronous LMs upon the diagnosis of primary cancer.[19]
LM is a major factor related to poor progression-free survival (PFS) and OS among patients with NSCLC.[4] For instance, a previous cohort study of 1096 patients with NSCLC and LMs conducted between 2006 and 2014 reported a 6-month survival rate of 68.2%.[20] The reported median OS of patients with and without LMs who received nivolumab treatment ranged from 3.6 to 7.5 months and 5.8 to 20.7 months, respectively.[21] Among patients with chemotherapy-naïve, advanced EGFR-mutated NSCLC without brain metastasis, the median PFS was markedly poorer in those with LMs than in those without LMs (5.1 months and 12.9 months, respectively).[22] A single-center study involving 41 patients reported immune checkpoint inhibitor (ICI) therapy response rates of 22.5% and 29.7% in patients with and without LMs, respectively (P = 0.43).[23]
Pathophysiological Mechanisms in LM of NSCLC Anatomical featuresThe liver receives its blood supply from two sources, namely two-thirds to three-fourths of the blood flows from the portal vein, while the remainder flows from the hepatic aortic system. The hepatic artery is the primary route by which tumor cells enter the liver,[24] while lung cancers seed tumor cells into the liver via the portal vein.[3] The specific anatomical localization, immunosuppressive environment, and endothelial cell infiltration of the liver make it susceptible to tumor metastasis from extrahepatic cancers, referred to as secondary liver cancers.[25] For example, the cluster of differentiation 8 positive (CD8+) T cells targeting antigens expressed in the liver may exhibit impaired function (T-cell failure) when continuously exposed to a high antigen load within the liver after their initial activation in the secondary lymphoid organs,[25] thus providing the immunosuppressive microenvironment for LMs.
Genomic aberrationsLM can arise from genomic events in the cancer cells, such as the activation of oncogenes and inactivation of tumor suppressor genes. For instance, USP22, a ubiquitin hydrolase, enhances the proliferation, angiogenesis, and recurrence of NSCLC.[26]USP22-knockout is a novel NSCLC treatment strategy that may activate a wide range of anticancer activities by affecting multiple signaling pathways that inhibit angiogenesis, proliferation, epithelial-to-mesenchymal transition (EMT), and the expression of KRAS and c-Myc.[26] In lung cancer metastasis models in vivo, USP22-knockout cancer cells generated substantially smaller and fewer LMs.[26]
The expression of 5′-nucleotidase domain-containing 2 (NT5DC2), an NT5DC family member, is increased in human NSCLC tissues.[27] Mechanistically, the proliferation and apoptosis of NSCLC are regulated by NT5DC2 in a p53-dependent manner. NT5DC2 suppression triggers both cell cycle arrest and apoptosis in NSCLC cells in the G2/M phase, while the deletion of NT5DC2 inhibits EMT and NSCLC-related LM.[27]
The inhibitor of differentiation (ID) family of proteins encourages tumor growth, angiogenesis, and metastasis.[28] ID1 is modulated via the c-Jun N-terminal kinase (JNK) pathway by KRAS oncogenes, and deletion of ID1 leads to the downregulation of mitogenic machinery elements through the suppression of the transcription factor FOS-like 1, together with several kinases in the KRAS signaling network. The expression of ID1 in the liver microenvironment and lung cancer cells promotes LM by increasing the colonization and migration ability of lung cancer cells, regulating the EMT phenotype, and establishing the premetastatic niche (PMN).[28] The abolition of ID1 induces G2/M phase arrest and apoptosis in KRAS-mutated LUAD.[27]Id1 deletion in xenograft models markedly compromises the growth and maintenance of tumors, together with LMs, thereby improving survival.[29]
Adenylate kinase 4 (AK4) is a biomarker in lung cancer metastasis,[30] the overexpression of which augments the hypoxia-inducible factor-1α (HIF-1α) protein expression by augmenting intracellular ROS levels and subsequently triggers EMT and invasion under hypoxic conditions. In a mouse model, AK4 overexpression reduces hypoxic necrosis and promotes LM.[30] Reducing ROS production with N-acetylcysteine eliminates the AK4-induced invasion potential under hypoxic conditions. Thus, the AK4–HIF-1α signaling axis might be a promising treatment target for the treatment of LM from lung cancer.
Collectively, the above-mentioned molecules, including USP22, ID1, NT5DC2, and AK4, can modulate the expression of proliferation-, angiogenesis-, and EMT-related proteins in either cancer cells or in the hepatic microenvironment. This modulation leads to the effective liver colonization of cancer cells and secondary metastatic outgrowth.[26–28,30,31] This finding underscores the potential of targeting EMT as a promising strategy for inhibiting LM.
Immunosuppressive microenvironmentsSeveral reliable preclinical models have been used to evaluate the hepatic metastasis of tumors, including implantation mouse models,[27,32–34]ex vivo liver model systems,[35–37] genetically engineered mice,[38] and patient-derived xenografts.[38] Although these models present both merits and shortcomings[39–41] [Supplementary Table 1, https://links.lww.com/CM9/B859], two key ecological niches promoting the LM process have been elucidated using a cellular and hepatic metastasis mouse model of NSCLC. First, the TME at secondary organ sites can favorably influence metastatic growth before tumor cell entry, which is termed the PMN. A PMN is created by molecules secreted from the primary tumor that recruit non-tumor cells such as KCs, HepSCs, myeloid-derived suppressor cells (MDSCs), endothelial cells, and neutrophils, as well as extracellular matrix molecules, including collagen I, II, and IV; FAT atypical cadherin 1 (FAT1); Wnt/β-catenin; S100 in the liver.[18] The PMN is formed early, even before tumor cells enter the liver.[42] Second, the post-invasive tumor ecological niches that develop after the entry of tumor cells can be described in four critical phases: the microvascular phase, which involves the invasion of cancer cells and their arrest in the sinusoidal vessels; the pre-angiogenic stage outside the vessels; the angiogenic phase that supplies cancer cells with nutrients and oxygen; and finally, the phase in which LMs expand and grow into clinically detectable tumors.[18] These various cell types, along with the growth factors and chemokines/cytokines they secrete and the molecular signaling involved, represent potential targets for preventing and treating LM [Figures 1 and 2].
 Figure 1:
Figure 1: Phases of NSCLC liver metastases. Liver metastases develop after the hematogenous spread of NSCLC cells from the lungs to the liver, which provides “fertile soil” to develop metastasis. First, tumor cells invade the surrounding tissues, venules, capillaries, lymphatic system (intravasation), and circulation, and survive in the circulation; subsequently, the tumor cells adhere to the vascular wall and extravasate from the hepatic vasculature. After extravasation, the tumor cells experience four pivotal phases, including the microvascular, pre-angiogenic, angiogenic, and growth phases, causing micrometastases; later colonize and form macroscopic liver metastases. MDSCs: Myeloid-derived suppressor cells; PMN: Premetastatic niche; NSCLC: Non-small cell lung cancer.
 Figure 2: Pro-metastatic and anti-metastatic microenvironment of the liver in NSCLC. (A) FGF9 promotes EMT by interacting with FGFR1 on Lewis lung cancer cells.[49] Steatosis-activated HepSCs promote angiogenesis and metastasis during LM development.[59] Hepatic monocyte-derived CD11b+F4/80+ macrophages lead to antigen-specific T-cell apoptosis through the Fas–FasL pathway.[4] MDSCs that generate immunosuppressive cytokines, such as TGF-β and IL-10, express arginase to inhibit the proliferation of T cells and ROS generation.[50] Hepatocytes activate transcriptional activator 3 signaling and signal transducers depending on IL-6.[55] Neutrophils promote cancer cell adhesion mediated by neutrophil Mac-1/ICAM-1 within liver sinusoids and facilitate LM.[43] The molecules and signaling pathways involved in LM include NT5DC2, USP22, NOX4, EGFR, FUT8, KRAS, ID1, and c-Myc. (B) Activated CD8+ T cells induce NSCLC cell apoptosis in the liver. Neutrophils prevent metastasis mediated by an accelerated neutrophil maturation rate and anti-metastatic activity.[44] NK cells present anti-metastatic tumor ability following IFN-γ induction.[54] Dectin-2 mediates the uptake and clearance of cancerous KCs.[48] CD: Cluster of differentiation; EGFR: Epidermal growth factor receptor; EMT: Epithelial-to-mesenchymal transition; FGF9: Fibroblast growth factor 9; FGFR1: Fibroblast growth factor receptor 1; FUT8: Fucosyltransferase 8; HepSCs: Hepatic stellate cells; Id: Inhibitor of differentiation; IFN-γ: Interferon-γ; ICAM-1: Intercellular adhesion molecule-1; IL: Interleukin; KCs: Kupffer cells; LM: Liver metastasis; Mac-1: Membrane-activated complex 1; MDSCs: Myeloid-derived suppressor cells; NK: Natural killer; NOX4: NADPH oxidase 4; NSCLC: Non-small cell lung cancer; NT5DC2: 5′-Nucleotidase domain containing 2; ROS: Reactive oxygen species; TGF-β: Transforming growth factor-β.Role of liver-specific cells
Figure 2: Pro-metastatic and anti-metastatic microenvironment of the liver in NSCLC. (A) FGF9 promotes EMT by interacting with FGFR1 on Lewis lung cancer cells.[49] Steatosis-activated HepSCs promote angiogenesis and metastasis during LM development.[59] Hepatic monocyte-derived CD11b+F4/80+ macrophages lead to antigen-specific T-cell apoptosis through the Fas–FasL pathway.[4] MDSCs that generate immunosuppressive cytokines, such as TGF-β and IL-10, express arginase to inhibit the proliferation of T cells and ROS generation.[50] Hepatocytes activate transcriptional activator 3 signaling and signal transducers depending on IL-6.[55] Neutrophils promote cancer cell adhesion mediated by neutrophil Mac-1/ICAM-1 within liver sinusoids and facilitate LM.[43] The molecules and signaling pathways involved in LM include NT5DC2, USP22, NOX4, EGFR, FUT8, KRAS, ID1, and c-Myc. (B) Activated CD8+ T cells induce NSCLC cell apoptosis in the liver. Neutrophils prevent metastasis mediated by an accelerated neutrophil maturation rate and anti-metastatic activity.[44] NK cells present anti-metastatic tumor ability following IFN-γ induction.[54] Dectin-2 mediates the uptake and clearance of cancerous KCs.[48] CD: Cluster of differentiation; EGFR: Epidermal growth factor receptor; EMT: Epithelial-to-mesenchymal transition; FGF9: Fibroblast growth factor 9; FGFR1: Fibroblast growth factor receptor 1; FUT8: Fucosyltransferase 8; HepSCs: Hepatic stellate cells; Id: Inhibitor of differentiation; IFN-γ: Interferon-γ; ICAM-1: Intercellular adhesion molecule-1; IL: Interleukin; KCs: Kupffer cells; LM: Liver metastasis; Mac-1: Membrane-activated complex 1; MDSCs: Myeloid-derived suppressor cells; NK: Natural killer; NOX4: NADPH oxidase 4; NSCLC: Non-small cell lung cancer; NT5DC2: 5′-Nucleotidase domain containing 2; ROS: Reactive oxygen species; TGF-β: Transforming growth factor-β.Role of liver-specific cells
LSECs, KCs, HepSCs, parenchymal hepatocytes, DCs, and resident natural killer (NK) cells all correlate with the promotion and maintenance of LMs, which mainly induce the expression of immune cells, such as neutrophils, macrophages, and monocytes, among others.[18]
NeutrophilsNeutrophils, which are part of an innate immune response to pathogens, can also be activated promptly by invading cancer cells. The number of neutrophils present in peripheral blood correlates positively with LM in patients with lung cancer.[43] In a murine model, the interaction between circulating tumor cells and adherent neutrophils in the inflamed sinusoids may be conducive to the arrest of Lewis lung carcinoma (LLC) cells in the liver.[44] Neutrophils promote cancer cell adhesion, mediated by neutrophil membrane-activated complex 1 (Mac-1)/intercellular adhesion molecule-1 (ICAM-1) within the liver sinusoids, thereby facilitating LM.[45] However, atypical chemokine receptors are leukocyte trafficking, immunity, and inflammation modulators.[46] In an orthotopic mouse model with 4T1 mammary carcinoma, ACKR2-deficient neutrophils showed an accelerated maturation rate, thus preventing neutrophil-mediated metastasis.[46] Therefore, neutrophils possess both anti- and pro-metastatic functions based on disease context.[47]
MacrophagesMacrophages are associated with liver metastasis from lung cancer. Chang et al[48] found that intratumor injections of fibroblast growth factor 9 (FGF9) promote EMT, infiltration of M2-macrophage into the TME, and LM via its interaction with fibroblast growth factor receptor 1 (FGFR1) in a subcutaneous LLC-bearing mouse model. FGF9 stimulates LM and LLC tumorigenesis by activating the extracellular signal-regulated kinase (ERK), protein kinase (AKT), and focal adhesion kinase (FAK) signaling via the FGFR1.
KCs account for approximately 10% of hepatocytes and are the largest tissue-resident macrophage population,[49] and critically mediate the immunosuppressive liver microenvironment through the expression of cytotoxic T-lymphocyte antigen-4 (CTLA-4), programmed cell death ligand 1 (PD-L1), transforming growth factor-β (TGF-β), or interleukin (IL)-10, and reduction of the levels of co-stimulatory molecules (CD80 and CD86), thereby activating Tregs.[50] Targeting KCs might be a promising strategy for preventing the development of incipient LMs. KC depletion by gadolinium chloride results in a decreased liver tumor burden. Dectin-2, a unique C-type lectin selectively expressed by DCs, mediates cancer cell uptake and clearance by KCs.[51] Kimura et al[51] reported that dectin-2-mediated cancer cell phagocytosis by KCs in an in vivo LUAD mouse model demonstrated LM suppression. Mice with LMs exhibited increased expression of CD11b+F4/80+ myeloid cells along with a decreased expression of CD4+ T cells. Mechanistically, the liver monocyte-derived CD11b+F4/80+ macrophages caused antigen-specific T-cell apoptosis via the FAS–Fas ligand (FASL) pathway.[4]
MDSCsAnother heterogenic immature myeloid cell with potent immunosuppressive function is the MDSC. MDSCs generate immunosuppressive cytokines, such as TGF-β and interleukin (IL)-10, and express arginase, which inhibits the proliferation of T cells and ROS generation.[52] Therefore, immunosuppressive myeloid cells, such as MDSCs and M2 macrophages functioning as main barriers against effector lymphocytes, are potential targets for immunomodulatory drugs.
T cellsThe presence of LMs is related to fewer CD8+ T cells at the margins of distant tumor infiltration, a cellular characteristic that correlates with a response to programmed cell death protein 1 and its ligand (PD-1)/PD-L1 inhibitors.[53] Eliminating antigen-specific CD8+ T cells results in systemic immunosuppression.[4] Recent studies examining the relationship between LM and treatment response in patients with lung cancer revealed a decreased density of CD8+ T cells at the margins of invasive tumors in LM biopsies, which may explain the poor survival.[53] Several mechanisms have been proposed to account for an immune tolerance-induced liver, such as the activation of Tregs by KCs.[54] Tregs suppress abnormal self-antigen and antitumor immune responses.[12]
DCsCompared with DCs from other tissues, liver DCs poorly affect T-cell activation due to their immaturity and the high IL-10 and low IL-12 levels in the hepatic microenvironment.[55] The hepatic microenvironment enables the differentiation of hematopoietic progenitor cells into regulatory DCs that sustain hepatic tolerance.[50]
NK cellsAs a heterogeneous population, hepatic resident NK cells exert an essential role in infection and tumor immunosurveillance. The liver participates in lung metastasis as a leukocyte supplier.[56] Liver-derived leukocytes display liver-like characteristics and are designated hepato-entrained leukocytes, which contain B220+CD11c+NK1.1+ cells with antimetastatic tumor ability following interferon-γ induction.
HepatocytesHepatocytes are the bases of the liver and comprise approximately 60% and 80% of the liver’s cell number and mass, respectively.[3] Although data related to the effect of hepatocytes on LM progression remain scarce, available studies have reported that hepatocytes coordinate the accumulation of bone marrow cells and fibrosis, helping to create a microenvironment conducive to metastasis.[57] In mice displaying early pancreatic tumorigenesis, hepatocytes activate the signal transducers and activators of transcription 3. This process depends on the release of non-malignant cell-derived IL-6 into the circulation and enhances the production of serum amyloid A1 and A2.[57]
LSECsAs the initial barrier to cancer cell invasion, LSECs are a source of cytokines for the recruitment of immune cells following activation by invading cancer cells.[58] The specific characteristics of the endothelial cell surface may play an essential role in determining metastatic patterns.[59] This finding may be attributed to the differences in tumor cell adhesion propensity along with the preference for various endothelial cells. Disruption of the interaction between tumor cell-derived selectin ligands and endothelial cell selection reduces the recruitment of LLC cells to the liver.[44]
HepSCsHepSCs account for approximately 15% of non-parenchymal hepatocytes and coordinate the fibrotic response of the liver (e.g., wound healing). Hepatic steatosis, which causes liver inflammation and fibrosis, is an individual predictor of LM in patients with NSCLC.[60] Mechanistically, steatosis-activated HepSCs impact the progression and promotion of metastasis and organize angiogenesis during LM development.[61]
Cell interaction mediated by cytokines and chemokinesGrowth factor and chemokine receptors are the most intriguing emerging targets in the TME as they are responsive to pharmacotherapy using antibody- and small molecule-based strategies. Preclinical data have supported their usefulness as therapeutic targets for LMs. Infectious complications have been associated with an increased risk of metastasis following tumor resection.[62] The innate immune system can be activated with lipopolysaccharide (LPS). Toll-like receptor 4 recognizes LPS, a gram-negative bacterial cell wall component. In C57BL/6 mice, gram-negative Escherichia coli-related pneumonia enhances LM in murine H59 NSCLC via the activation of Toll-like receptor 4.[62]
TGF-β, produced by tumor cells or the surrounding stromal cells,[63] is a primary driver of the fibrotic and immunosuppressive microenvironment and is critical for angiogenesis and growth of LM.[64] TGF-β1 bound to the membranes of MDSCs is the primary negative regulator of hepatic NK cells, inhibits the proliferation of T cells, and induces Tregs in tumor-bearing hosts.[52] TGF-β signaling by monocyte myeloid cells inhibits CD8+ T cell activity during lung metastasis and promotes tumor growth in LM.[65] Cells pretreated with TGF-β1 tend to metastasize to the liver in experimental lung cancer metastasis mouse models.[66] Meanwhile, blocking TGF-β triggers a potent and lasting cytotoxic T-cell activity against cancer cells and prevents metastasis.[67] Therefore, TGF-β is one of the central cytokines in LM,[68] and future studies should focus on investigating TGF-β inhibitory strategies.
The complex interplay of chemokine ligand–receptors among different cells further complicates the signal transduction cascade in LM, inducing tumor angiogenesis, EMT, and migration/invasion.[69] For instance, tumor-derived chemokines C-C motif chemokine ligand 5 (CCL5) and CCL7, as type IV collagen regulatory genes, facilitate LM of lung carcinoma (M27colIV) cells via FAK signaling.[70] Autocrine CCL7/C-C motif chemokine receptor 3 (CCR3) signaling functions downstream of collagen IV/integrin α2/FAK signaling and promotes EMT and cancer cell migration. This signaling is conveyed via the MEK/ERK and phosphoinositide 3-kinase (PI3K) pathways and involves nuclear factor kappa B (NF-κB) activation downstream of AKT. Moreover, CCL5/CCR5 signaling regulates the T-cell response in the TME.[70]
Cell–cell communication mediated by exosomal substances in LMsExosomes, a subset of extracellular vesicles (EVs), contribute to building a supportive microenvironment for LMs[71] and contain proteins, microRNAs (miRNAs), long non-coding RNAs (lncRNA), and messenger RNA (mRNAs).[71] Jiang et al[34] demonstrated that the overexpression of the lncRNA ALAHM in EVs derived from LUAD cells markedly facilitates LM in a murine lung cancer model. LUAD-derived ALAHM in EVs also promotes hepatocellular secretion of hepatocyte growth factor by binding to AUF1. This event enhances LUAD cell proliferation, invasion, and migration. Consequently, ALAHM-containing EVs derived from LUAD cells increase the hepatocyte growth factor levels and promote LM of LUAD cells.
Additionally, a distinct correlation was found between LM and serum exosomal miRNAs in NSCLC.[72] miRNAs (e.g., miR-198-5p) impact the formation of mouse LMs by directly targeting fucosyltransferase 8 (FUT8) in NSCLC in vivo.[73] Lung cancer cell-derived exosomes containing miR-122-5p induce the EMT-related mechanisms in hepatocytes. These mechanisms include N-cadherin and vimentin expression enhancement, reduction of the expression of epithelial marker E-cadherin, and induction of liver cell migration, thereby establishing a pre-metastatic microenvironment for metastatic lung cancer.[74] Moreover, bone marrow-derived cell-secreted EVs containing miR-92a promote LM of lung cancer by potentiating HepSC activation, subsequently increasing extracellular matrix deposition in mice. This environment promotes the formation of an immunosuppressive PMN for LM by boosting the recruitment of MDSCs.[75] Mechanistically, miR-92a in EVs augments the activation of HepSCs induced via TGF-β signaling, by directly targeting the suppressor of mothers against decapentaplegic family member.
Overall, the liver microenvironment consists of various cell types, including neutrophils, macrophages, MDSCs, T cells, DCs, NK cells, and other cells. The above cell types interact with each other via cytokines/chemokines, including CCL7/CCR3, CCL5/CCR5, and TGF-β, and exosomal substances, i.e., lncRNAs and miRNAs [Figure 2]. Together, these impact the proliferation, invasion, migration, and EMT of NSCLC cells, thus influencing the establishment and growth of metastases. They represent potential targets for preventing and treating LM. However, LM is a complex process involving multiple factors. Despite the available data, liver metastatic TME is incompletely understood due to its complexity and heterogeneity. Studies to date have only preliminarily explored the underlying mechanism. An in-depth and extensive understanding of the specific mechanism using novel technologies, such as single-cell sequencing, spatial profiling technologies, and multiplex immunofluorescence, is required in future preclinical and clinical studies with large sample sizes.
Prospective Preclinical and Clinical Strategies for Targeting the Liver Metastatic MicroenvironmentTME is a key factor influencing tumor treatment outcomes. LMs can promote systemic tolerance of antigen-specific T cells via immunosuppressive TME in preclinical animal models and cancer patients.[4] Different immunosuppressive phenotypes in the liver microenvironment are potential therapeutic targets in patients with LMs.[12] Although clinical data have demonstrated the therapeutic effect of immunotherapy coupled with chemotherapy, radiotherapy, and vascular endothelial growth factor (VEGF) blockade for targeting the liver TME [Table 1], limited preclinical findings illustrate the implication of immunotherapy coupled with radiotherapy and neutrophil extracellular traps (NETs) inhibitors in lung cancer patients with LMs [Table 2].
Table 1 - Therapeutic agents for liver metastases of NSCLC in clinical trials. Therapeutic strategies Therapeutic agents Condition/population Phase Results Population (LMs/all) Study name [NCT No.] Refs
留言 (0)