
記住我
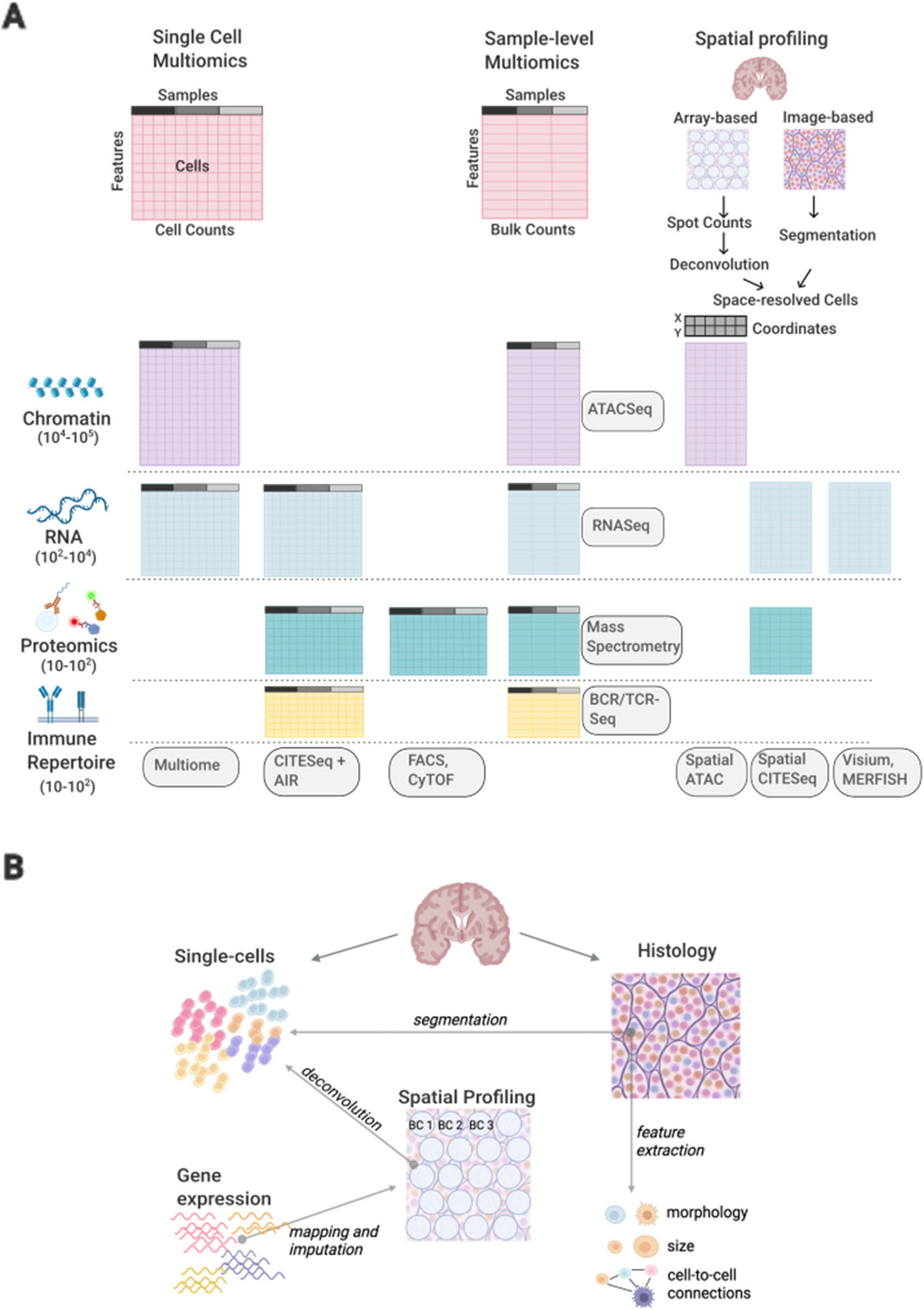
The primary objective of imply is to improve the accuracy of cell abundance estimations through the usage of a “subject- and cell-type specific reference panel”, which is a personalized CTS reference panel unique to each study participant. The algorithm is structured into three stages. In Stage I, the initial cell type compositions will be estimated using a population-level CTS reference panel. This first stage is very much alike existing deconvolution frameworks, but provides a valid initial estimation for downstream stages. The core component of imply lies in Stage II, where we optimize the usage of repeatedly-measured samples within each subject. Multi-measured samples, within each subject, are assumed to share the same reference panel but have cell type composition variations. Here, mixed-effect modeling is naturally adopted to capture the group-level average (fixed effect) and subject-level deviations (random effect). The output from this stage, for each subject, is a personalized reference panel. In Stage III, the personalized deconvolution can be easily conducted by adopting the personalized reference panel from Stage II, for each subject.
Model notationsWe use g to index the features (e.g., genes), where \(g=1,2,3,\dots , G\), and n to index the study subjects, where \(n=1,2,3,\dots , N\). For each subject n, the repeated or longitudinal samples are indexed by i, where \(i=1,2,\dots , t_n\). For each subject n, the observed bulk transcriptome dataset can be represented by \(\varvec\) below, which is of dimension \(G \times t_n\):
$$\begin \varvec=\left[ \begin y_ & y_ &\dots & y_ \\ y_ & y_ &\dots & y_ \\ \vdots & \vdots & y_ &\vdots \\ y_ & y_ &\dots & y_ \end \right] \end$$
Here, each element \(y_\) in \(\varvec\) has three indexes: g to index the feature, n to index the subject, and i to index the sample. The whole bulk transcriptome dataset from all subjects, which can be represented as \(\varvec = (\varvec, \varvec, \cdots , \varvec)\), is of dimension \(G \times T\). Here, T is the total number of samples across N subjects, and thus \(T= \sum _^t_n\). We use k to index cell types, where \(k=1,2,\dots , K\).
Stage I: Initial cell type proportion estimationInitially, similar to existing deconvolution methods, it is easy to obtain a single reference panel \(\varvec\) of dimension \(J \times K(J < G)\) for the study population, where J indicates the total number of usable and discriminative signature genes for deconvolution. Previous studies have demonstrated the feasibility of constructing a reference panel from pure cell line data or annotated single-cell RNA-seq (scRNA-seq) data [44, 49], and thus \(\varvec\) can be treated as known. With the observed bulk data \(\varvec\) and the initial reference panel \(\varvec\), as illustrated in the top-left of Fig. 1, the first-round reference-based ‘coarse’ deconvolution is conducted using a \(\nu\)-Support Vector Regression algorithm (\(\nu\)-SVR) based on a linearity assumption. Such strategy was already proven to be a successful choice in leading deconvolution algorithms. To be specific, this stage requires both the signature matrix \(\varvec\) and the feature-overlapped RNA-sequencing data \(\varvec\), comprising only the overlapping features filtered by marker genes from the signature matrix. For each subject n and sample i, the deconvolution is thus a regression problem: \(\varvec_ = \varvec \varvec_ + \varvec\), where \(\varvec\in R^J\) is the error term. Our initial deconvolution parameter-of-interest is \(\varvec_\), and can be estimated by minimizing the following objective function:
$$\begin \frac \Vert \varvec_ \Vert ^2 + C\sum \limits _^, \ \ \xi _j, \xi _j^* >0 \end$$
The solved \(\hat}_=\left(\hat_, \hat_,\dots \hat_\right)'\), for each sample, is a cell type abundance vector of dimension \(K \times 1\). The constraints of the objective function and parameters of \(\epsilon\), C, \(\xi _j\), and \(\xi _j^*\) are detailed in the Additional file 1: Method Details. Negative estimates in \(\hat}_\) are set to 0, and the remaining coefficients are normalized to sum-to-one, which is the general practice in proportion deconvolution. Repeating this process for all T samples across all subjects, we obtain the deconvoluted cell compositions. It is worth noting that although this deconvolution stage has little difference from existing methods, it provides a valid initial estimation for downstream steps.
Stage II: Personalized reference panel recoveryIn this stage, the inputs are the original bulk transcriptome data \(\varvec\), and the solved cell type compositions \(\hat}_\), for all samples from subject n. The goal is to solve for “subject- and cell-type-specific” reference panels. The key is to optimize the usage of repeatedly-measured samples within each subject. In this stage, we make an assumption that the multi-measured samples, within each subject, would share the same CTS reference panel. In other words, the transcriptome variations in observed bulk samples, within each subject, are primarily caused by cell type composition discrepancies. This is a moderate assumption, considering the compositional nature of multiple samples from the same tissue (for example, samples from multiple regions per brain). Here, mixed-effect regression would be a natural choice to capture the group-wise transcriptome average (fixed effect) and subject-level deviations (random effect) from the group average. Such modeling also allows for the consideration of additional covariates (Additional file 1: Method Details). Using the original bulk transcriptome data \(\varvec\) and the cell-type-specific and sample-specific compositions \(\hat_\) from Stage I, the following linear mixed-effect regression can be formulated for each gene g. Here, we drop the gene index g to simplify notation, but note this framework can be applied in parallel to all genes-of-interest to solve for “subject- and cell-type-specific” references.
$$\begin E(y_) = \sum \limits _^K \left( m_+ \beta _kz_n +u_\right) \hat_ \end$$
Here, the known independent variables are a group label \(z_n\) for each subject n, and estimated cell type compositions \(\hat_\) from Stage I for all samples. The coefficients-of-interest include group-level fixed effects \(m_\), \(\beta _k\), and subject-level random effect \(u_\). The interpretation is straightforward: \(m_\) is the average gene expression level in cell type k for the control group (\(z_n=0\)), and \(m_+\beta _k\) is the average gene expression in cell type k for the case group (\(z_n=1\)). Apparently, \(\beta _k\) is the differential expression across the two groups for a cell type k. Most importantly, the random effect \(u_\) represents a subject-specific deviation from the group-wise mean expression in cell type k. Note this modeling example above reflects the most basic scenario where study subjects originate from two groups (for example, cancer versus normal), where a binary scalar \(z_n\) is adopted to indicate group labels. This modeling can be extended naturally to incorporate additional covariates, either at the cell-type level or the subject-level. Modeling details and design matrices setup specified in the Additional file 1: Method Details. \(\hat_k\), \(\hat_k\) and \(\hat_\) are obtained by penalized least square algorithm with restricted maximum likelihood. The subject- and cell-type-specific reference panel is obtained by addition (fixed effect + random effect), with respect to each corresponding condition, cell type, and subject. To be specific, for subject n and cell type k, its purified reference expression is \(r_ = \hat_+z_n\hat_ + \hat_\). After repeating the same model for all G genes and adding gene index g back to \(r_\), the personalized reference panel for each subject n can be represented by a matrix \(\varvec_n\) of dimension \(G\times K\):
$$\begin \varvec_n = \left[ \begin r_ &r_ & \dots & r_ \\ r_ &r_ & \dots & r_ \\ \vdots & & \ddots & \\ r_ &r_ & \dots & r_ \end\right] \end$$
Stage III: Personalized deconvolutionWith the personalized reference panel \(\varvec_n\) available for each subject n, and the original bulk mixture transcriptome data, as shown in the lower-right corner of Fig. 1, we use non-negative least squares to deconvolute the cell type abundance \(\varvec_\). Here, we solve for \(\varvec_\) by optimizing the following objective function, for each subject n, under the constraint \(\varvec_ \ge 0\):
$$\begin \Vert \varvec_n \otimes \varvec_ vec\left(\varvec'_\right)- vec\left(\varvec'_\right) \Vert _2 \end$$
\(\varvec_\) is of dimension \(K\times t_n\) for each subject n. This is a joint optimization across all the samples per subject simultaneously instead of sample-wise optimization, using the subject-specific signature matrix \(\varvec_n\) and quadratic programming. The subscript I stands for the imply-estimated cell type abundance, in contrast to the coarse deconvolution abundance \(\varvec_\) from Stage I. Note that \(\nu\)-SVR with sample-wise optimization can also be utilized in this stage as an alternative approach, and we name this variant as imply-s. Overall, instead of using the population-level signature matrix \(\varvec\), the key of imply is to adopt a personalized \(\varvec_n\) to serve in the cell type abundance inference.
SimulationsPure cell-type-specific expression profilesNotations of gene g, subject n, sample i, and cell type k are borrowed from the previous section. The simulation scheme is borrowed and adapted from on our prior benchmark study [39], offering a comprehensive and flexible simulation framework. We utilized a set of true cell line RNA-seq dataset [34] to obtain the distribution of gene expression parameters in a genome-wide scale. This study has six immune cell types (neutrophils, monocytes, B-cells, CD4 T cells, CD8 T cells, and natural killer cells). For each cell type, the CTS gene expression parameters, expression means (\(\mu _\)) and biological dispersion (\(\phi _\)), are obtained by using the PROPER [53] package. There are correlations across cell type for both expression means and dispersion, as expected. Therefore, for the reference panel simulation, we use Multivariate Normal Distribution (MVN) to capture correlations for both expression mean and dispersion, in the log scale. We use \(\hat}_\) (\(\bar}_m\)) and \(\hat}_\) (\(\bar}_\phi\)) to denote variance-covariance matrices of expression mean and dispersion, respectively. The dimensions match the number of cell types and the details of \(\hat}_\), \(\bar}_m\), \(\hat}_\), and \(\bar}_\phi\) can be found in the Additional file 1: Simulation Details. We conduct 30 iterations for each simulation scenario, with six cell types and 1,000 genes:
$$\begin \varvec & \sim MVN\left(\bar}_m,\hat}_\right)\\ \varvec & \sim MVN\left(\bar}_\phi ,\hat}_\right) \end$$
Note that the mean expression \(\varvec\), and the biological dispersion \(\varvec\) are still parameter matrices for downstream usage. The case and control groups share the same \(\varvec\), but distinct mean expressions. The effect size of differential expression is defined by Log-Fold-Changes (LFC) denoted by \(\Delta\). The means for control and case are denoted by \(\varvec_=\varvec\) and \(\varvec_=\varvec+\Delta\). We introduce 10% of differentially expressed (DE) genes on cell types 1, 2, 3, and 4, respectively. The true CTS gene expression matrix \(\varvec\) is derived from a Gamma Distribution for both case and control:
$$\begin \varvec_ \sim \Gamma \left( \frac, \exp (\varvec_) \times \exp (\varvec) \right) \end$$
Subject-to-subject variations (SSV) are also introduced, implemented as the expression change percentages over the baseline in \(\varvec_\). Variations are then added to \(\varvec_\) to obtain subject-specific underlying gene expression matrices \(\varvec_n\). To reflect the various levels of variations, the level of SSV can take the following ranges: 0-5%, 5%-10%, 10%-20%, and 20%-50%. The total subject count per case/control group can take value in 25, 50, 75, and 100. The subject-level cell-type-specific underlying gene expression is shared across multiple samples, and each subject is measured 3 times.
Fig. 2
imply can improve cell type deconvolution accuracy. A Scatterplot showing imply estimated gene expression reference panel versus the true reference panel values. B Superimposed scatterplot of the imply-estimated cell type proportion over the CIBERSORT-estimates, which are the results from the current state-of-art method. imply shows better concordance with the ground truth. C-F Boxplots displaying evaluation metrics and each point representing one simulation iteration: ABD, rABD, CD, and \(\Delta \rho _}\). Five additional modeling frameworks are benchmarked. The red dashed line (value of 0) represents no improvement in proportion estimation. For (C) and (D), lower values indicate better deconvolution accuracy. For (E) and (F), higher the better
Cell type proportions and observed read countsTo generate the cell type proportions, we borrow information from multiple well-labeled single cell RNA-seq studies. We mix and bootstrap cell labels from a combined pool and obtain the empirical cell proportions from this resampling. We use Dirichlet Distribution to estimate \(\varvec\) parameters and simulate cell type proportions. The detailed procedures for generating cell proportions are outlined in the Additional file 1: Simulation Details. The simulated sample-specific cell proportions are:
$$\begin \varvec} \sim Dirichlet(\varvec_) \end$$
\(\varvec}\) are reorganized into cell composition matrix, \(\varvec_T\). The sample-specific underlying gene expression reference panel is the weighted average across cell types in \(\varvec_n\) by \(\varvec_\), denoted as \(\varvec_ = \varvec_n\times \varvec_^\), and will follow a Gamma Distribution as well [41]. \(\varvec_\) is further assessed by the Poisson Distribution to generate observed RNA-sequencing counts data, denoted as:
$$\begin \varvec_ \sim Pois(\varvec_), \end$$
for subject n at measurement i across all G genes. Overall, the Gamma Distribution models biological variations, the Dirichlet Distribution regulates cell type proportion variations, and Poisson Distribution mimics technical noise related to the randomness in the sequencing experiments. This multi-step simulation design enables the separation of biological and technical noise [16, 39], among other factors, to facilitate a comprehensive simulation study for our model testing.
$$\begin \varvec_T= \left[ \begin \theta _ &\theta _&\dots & \theta _\\ \theta _ &\theta _&\dots & \theta _\\ \vdots & & \ddots & \\ \theta _1} &\theta _2}&\dots & \theta _K}\\ \vdots & & \ddots & \\ \theta _ &\theta _&\dots & \theta _\\ \theta _ &\theta _&\dots & \theta _\\ \vdots & & \ddots & \\ \theta _1} &\theta _2}&\dots & \theta _K} \end\right] \end$$
Input signature matrixThe signature matrix is required by the algorithm as an input. To obtain it, we first take the average across all \(\varvec_n\) matrices to get CTS gene expression mean matrix. Then 300 or 500 pseudo-marker genes are selected by findRefinx function (ordered by coefficients of variation) from TOAST [32] to establish a signature matrix as the input for imply.
Evaluation metricsWe use \(\varvec\) and \(\hat}\) to denote the ground truth and estimated cellular abundances, which has the unique property of unit-sum and bounded by zero and one. Naturally, a central goal here is to assess how good the cellular abundances estimator \(\hat}\) is. Specifically, we denote imply’s deconvolution values as \(\hat}_I\), and existing method’s deconvolution results as \(\hat}_E\). The existing methods include currently available deconvolution approaches and those do not consider personalized reference panels. The following evaluation metrics are adopted for benchmarking:
Absolute bias differences (ABD) and relative absolute bias differences (rABD)
$$\begin ABD & := \sum | \hat}_I - \varvec | - \sum | \hat}_E - \varvec |, \\ rABD & := \left[ \varvec\left( \frac}_I - \varvec|}}\right) - \varvec\left( \frac}_E - \varvec|}}\right) \right] \times 100\% \end$$
Here, for both ABD and rABD, if they are smaller than zero, it means the imply successfully reduces the estimation bias. A smaller value further indicates better performance.
Correlation differences (CD)
$$\begin CD := corr\left( \hat}_I,\varvec\right) - corr\left( \hat}_E,\varvec\right) \end$$
Here, if CD>0, then imply increases the correlation between the estimation and the ground truth. A larger value indicates favorable performance.
Lin’s concordance correlation coefficient (CCC) and its variationsLin’s concordance correlation coefficient (Lin’s CCC) [31], denoted as \(\rho _}\), has been extensively used to evaluate the concordance between a new measure and a gold standard measurement, and is defined as:
$$\begin \rho _} \left( \varvec, \hat}\right) = 1-\frac - \hat}\right) ^2\right] } - \hat}\right) ^2\right] }, \end$$
where \(E_I\) indicates the expectation under the assumption that \(\varvec\) and \(\hat}\) are independent. Lin’s CCC is bounded between 1 (perfect agreement) and -1 (disagreement), and the concordance improves as \(\rho _} (\varvec, \hat})\) approaches 1. Additionally, we adopt a Euclidean distance-based variation of Lin’s CCC, by substituting the expected squared difference to Euclidean distance, denoted as \(\rho _}\), defined below:
$$\begin \rho _} \left( \varvec, \hat}\right) = 1-\frac^ \left( \varvec^ - \hat}^\right) ^2\right] }^ \left( \varvec^ - \hat}^\right) ^2\right] } \end$$
Another option is to employ the Aitchison [2] distance-based Concordance Correlation Coefficient (CCC), which is explained in detail in the Additional file 1: Evaluation Metric, with the results provided in the Additional file 1: Simulation Results. These metrics are adopted because they have been shown to be statistically more rigorous in dependent measures that are subject to the positiveness and unit-sum constraints [13], as is often the case in compositional proportion outcome. If imply yields increased concordance and improved precision, we would expect positive values in the differences of CCC. These metrics are respectively defined below:
$$\begin \Delta \rho _} & = \rho _} \left( \varvec, \hat}_I\right) - \rho _} \left( \varvec, \hat}_E\right) \\ \Delta \rho _} & = \rho _} \left( \varvec, \hat}_I\right) - \rho _} \left( \varvec, \hat}_E\right) \end$$
Overview of the PDBP and TEDDY cohortsReal data analysis was conducted on two cohorts: the Parkinson’s disease Biomarker Program (PDBP) and The Environmental Determinants of Diabetes in the Young (TEDDY) [29]. The PDBP consortium has the repeatedly measured RNA-seq datasets, demographic and clinical information collected from patients with or without Parkinson’s Disease (PD) recruited from multiple medical centers and research institutions in the United States between November 2012 and August 2018. The PDBP cohort data were collected longitudinally overtime for each subject, allowing us to track changes in cell type composition and disease progression over time. In our study de-identified participants with at least three observations over time were retained. A total of 399 PD patients and 173 controls, with 2599 longitudinal samples over 2 years, were included. Longitudinal RNA samples in PDBP were extracted from the whole blood. Clinical data includes information about patients’ medical history, symptoms, disease status, total Montreal Cognitive Assessment (MoCA) scores, and MDS UPDRS part III motor scores. The TEDDY cohort is a multi-center pediatric study of Type 1 diabetes (T1D). TEDDY cohort screened and enrolled participants with susceptibility of T1D based on the Human Leukocyte Antigen (HLA) genotypes from six clinical centers in four countries (U.S., Finland, Germany, and Sweden). A total of 8,676 high-risk infants were enrolled from birth and followed every 3 months for blood sample collection and islet autoantibody (IAbs) measurement up to 4 years of age. Details of sample collection, RNA sequencing procedures, bioinformatics processing, and quality control are described in the Additional file 1: Method Details and [54]. The longitudinal whole blood transcriptome data enable the imply deconvolution.
留言 (0)