
記住我


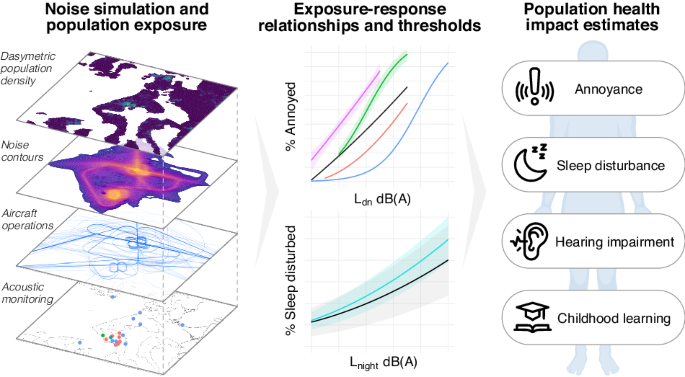

ABIS, a general population cohort consisting of 17,000 children born 1st of Oct 1997–1st of Oct 1999, followed prospectively with regular follow-ups. ABIS is connected to the Swedish National Diagnosis Register which give information about diagnosis of autoimmune disease. Stool samples were collected from ca 1800 individuals at 1 year of age, and microbiome studies have been performed [34, 39]. The present study includes subjects (N = 62) from this group who later developed one or more autoimmune and inflammatory diseases such as CD (N = 28), IBD (N = 7), hypothyroidism (HT) (N = 6), juvenile idiopathic arthritis (JIA) (N = 9) and T1D (N = 12), along with their controls matched for sex and age at the time of diagnosis (N = 268). The average age of diagnosis was 11.5 years in CD, 16 years in IBD, 15 years in JIA, 16 years in HT, and 13.5 years in T1D group [39]. The cohort is representative of a general population in Sweden. Related to socioeconomical factors (e.g., parental education) or other lifestyle factors, there was no statistically significant differences between the groups. The cord blood samples collected during birth were subjected to metabolomics analysis. Figure 1 summarizes the study design and integrated workflow. For more detailed information about demographic characteristics and individual diseases, readers are referred to our previous study [39].
Fig. 1: Summary of our work, which aimed to investigate how prenatal exposure to environmental contaminants alters the cord serum metabolome in the ABIS cohort.
We used metabolomics to determine the levels of exposure to environmental contaminants and metabolites in the cord blood. Our work involved three main stages. Firstly, we examined the levels of exposure and significant differences between control (N = 268) and cases (N = 62). Secondly, we studied the associations of contaminant exposure with cord serum metabolic profiles. Finally, we investigated the impact of contaminant exposure on cord serum metabolites. Overall, our work sheds light on the effects of environmental contaminants on the cord serum metabolome, which may have implications for the future progression of autoimmune diseases.
Analysis of metabolome and environmental contaminantsA total of 330 cord blood samples were randomized and analyzed as described below. Shortly, two methods were applied for separate extraction of lipids and polar/semipolar metabolites and the extracts were then analyzed using an ultra-high-performance liquid chromatography quadrupole time-of-flight mass spectrometry (UHPLC-QTOFMS) as described previously [39] and the data were processed using MZmine 2.53 [45]. Quantification was performed using calibration curves and the identification was done with a custom database. Level 1 identification refers to identified compounds where reference compounds are available, while level 2 identification denotes compounds identified based on their MS/MS in comparison with mass spectrometry libraries, as defined by the Metabolomics Standards Initiative (MSI). Quality control was performed by analyzing pooled quality control samples. In addition, extracted blank samples, standards compounds, and reference plasma (NIST SRM 1950); purchased from the National Institute of Standards and Technology at the US Department of Commerce (Washington, DC, USA)), were analyzed as part of the quality control procedure.
Analysis of molecular lipids10 µl of serum was mixed with 10 µl 0.9% NaCl and extracted with 120 µl of CHCl3: MeOH (2:1, v/v) solvent mixture containing internal standard mixture (c = 2.5 µg/ml; 1,2-diheptadecanoyl-sn-glycero-3-phosphoethanolamine (PE(17:0/17:0)), N-heptadecanoyl-D-erythro-sphingosylphosphorylcholine (SM(d18:1/17:0)), N-heptadecanoyl-D-erythro-sphingosine (Cer(d18:1/17:0)), 1,2-diheptadecanoyl-sn-glycero-3-phosphocholine (PC(17:0/17:0)), 1-heptadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine (LPC(17:0)) and 1-palmitoyl-d31-2-oleoyl-sn-glycero-3-phosphocholine (PC(16:0/d31/18:1)) and, triheptadecanoylglycerol (TG(17:0/17:0/17:0)). The samples were vortexed and let stand on the ice for 30 min before centrifugation (9400 rcf, 3 min). 60 µl of the lower layer of was collected and diluted with 60 µl of CHCl3: MeOH. The samples were kept at -80 °C until analysis.
Samples were analyzed by UHPLC-QTOFMS (Agilent Technologies; Santa Clara, CA, USA). The analysis was carried out on an ACQUITY UPLC BEH C18 column (2.1 mm × 100 mm, particle size 1.7 μm) by Waters (Milford, USA). The eluent system consisted of (A) 10 mM NH4Ac in H2O and 0.1% formic acid and (B) 10 mM NH4Ac in acetonitrile (ACN): isopropanol (IPA) (1:1) and 0.1% formic acid. The gradient was as follows: 0–2 min, 35% solvent B; 2–7 min, 80% solvent B; 7–14 min 100% solvent B. The flow rate was 0.4 ml/min.
The following steps were applied in data processing with MZmine 2.53: (i) Mass detection with a noise level of 1000, (ii) Chromatogram builder with a minimum time span of 0.08 min, minimum height of 1000 and a m/z tolerance of 0.006 m/z or 10.0 ppm, (iii) Chromatogram deconvolution using the local minimum search algorithm with a 70% chromatographic threshold, 0.05 min minimum RT range, 5% minimum relative height, 1200 minimum absolute height, a minimum ration of peak top/edge of 1.2 and a peak duration range of 0.08–5.0, (iv), Isotopic peak grouper with a m/z tolerance of 5.0 ppm, RT tolerance of 0.05 min, maximum charge of 2 and with the most intense isotope set as the representative isotope, (v) Join aligner with a m/z tolerance of 0.009 or 10.0 ppm and a weight for of 2, a RT tolerance of 0.15 min and a weight of 1 and with no requirement of charge state or ID and no comparison of isotope pattern, (vi) Peak list row filter with a minimum of 10% of the samples (vii) Gap filling using the same RT and m/z range gap filler algorithm with an m/z tolerance of 0.009 m/z or 11.0 ppm, (vii) Identification of lipids using a custom database search with an m/z tolerance of 0.008 m/z or 8.0 ppm and a RT tolerance of 0.25 min. Identification of lipids was based on an in-house library based on LC-MS/MS data on retention time and mass spectra. The identification was done with a custom database, with identification levels 1 and 2, i.e., based on authentic standard compounds (level 1) or based on MS/MS identification (level 2).
Quantification of lipids was performed using a 7-point internal calibration curve (0.1–5 µg/mL) using the following lipid-class specific authentic standards: using 1-hexadecyl-2-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine (PC(16:0e/18:1(9Z))), 1-(1Z-octadecenyl)-2-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine (PC(18:0p/18:1(9Z))), 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine (LPC(18:0)), 1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine (LPC(18:1)), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (PE(16:0/18:1)), 1-(1Z-octadecenyl)-2-docosahexaenoyl-sn-glycero-3-phosphocholine (PC(18:0p/22:6)) and 1-stearoyl-2-linoleoyl-sn-glycerol (DG(18:0/18:2)), 1-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine (LPE(18:1)), N-(9Z-octadecenoyl)-sphinganine (Cer(d18:0/18:1(9Z))), 1-hexadecyl-2-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine (PE(16:0/18:1)) from Avanti Polar Lipids, 1-Palmitoyl-2-Hydroxy-sn-Glycero-3-Phosphatidylcholine (LPC(16:0)), 1,2,3 trihexadecanoalglycerol (TG(16:0/16:0/16:0)), 1,2,3-trioctadecanoylglycerol (TG(18:0/18:0/18:)) and 3β-hydroxy-5-cholestene-3-stearate (ChoE(18:0)), 3β-Hydroxy-5-cholestene-3-linoleate (ChoE(18:2)) from Larodan, were prepared to the following concentration levels: 100, 500, 1000, 1500, 2000 and 2500 ng/mL (in CHCl3:MeOH, 2:1, v/v) including 1250 ng/mL of each internal standard. For unknown lipids, the results are given as normalized peak areas, after normalization with the closest eluting internal standard.
Analysis of polar metabolites40 µl of serum sample was mixed with 90 µl of cold MeOH/H2O (1:1, v/v) containing the internal standard mixture (Valine-d8, Glutamic acid-d5, Succinic acid-d4, Heptadecanoic acid, Lactic acid-d3, Citric acid-d4. 3-Hydroxybutyric acid-d4, Arginine-d7, Tryptophan-d5, Glutamine-d5, each at at c = 1 µgmL−1 and 1-D4-CA,1-D4-CDCA,1-D4-DCA,1-D4-GCA,1-D4-GCDCA,1-D4-GLCA,1-D4-GUDCA,1-D4-LCA,1-D4-TCA, 1-D4-UDCA, each at 0.2 1 µgmL−1) for protein precipitation. The tube was vortexed and ultrasonicated for 3 min, followed by centrifugation (10000 rpm, 5 min). After centrifuging, 90 µl of the upper layer of the solution was transferred to the LC vial and evaporated under the nitrogen gas to dryness. After drying, the sample was reconstituted into 60 µl of MeOH: H2O (70:30).
Analyses were performed on an Agilent 1290 Infinity LC system coupled with 6545 QTOFMS interfaced with a dual jet stream electrospray (dual ESI) ion source (Agilent Technologies; Santa Clara, CA, USA) was used for the analysis. Aliquots of 10 μL of samples were injected into the Acquity UPLC BEH C18 2.1 mm × 100 mm, 1.7-μm column (Waters Corporation, Wexford, Ireland), fitted with a C18 precolumn (Waters Corporation, Wexford, Ireland). The mobile phases consisted of (A) 2 mM NH4Ac in H2O: MeOH (7:3) and (B) 2 mM NH4Ac in MeOH. The flow rate was set at 0.4 mLmin−1 with the elution gradient as follows: 0-1.5 min, mobile phase B was increased from 5% to 30%; 1.5–4.5 min, mobile phase B increased to 70%; 4.5–7.5 min, mobile phase B increased to 100% and held for 5.5 min. A post-time of 5 min was used to regain the initial conditions for the next analysis. The total run time per sample was 20 min. The dual ESI ionization source settings were as follows: capillary voltage was 4.5 kV, nozzle voltage 1500 V, N2 pressure in the nebulized was 21 psi and the N2 flow rate and temperature as sheath gas was 11 Lmin−1 and 379 °C, respectively. In order to obtain accurate mass spectra in MS scan, the m/z range was set to 100-1700 in negative ion mode. MassHunter B.06.01 software (Agilent Technologies; Santa Clara, CA, USA) was used for all data acquisition.
MS data processing was performed using the same parameters as in lipidomic analysis.
Quantitation was done using 6-point calibration (PFOA c = 3.75–120 ng/mL, bile acids c = 20–640 ng/mL, polar metabolites c = 0.1 to 80 μg/mL). Quantification of other bile acids was done using the following compounds: chenodeoxycholic acid (CDCA), cholic acid (CA), deoxycholic acid (DCA), glycochenodeoxycholic acid (GCDCA), glycocholic acid (GCA), glycodehydrocholic acid (GDCA), glycodeoxycholic acid (GDCA), glycohyocholic acid (GHCA), glycohyodeoxycholic acid (GHDCA), glycolitocholic acid (GLCA), glycoursodeoxycholic acid (GUDCA), hyocholic acid (HCA), hyodeoxycholic acid (HDCA), litocholic acid (LCA), alpha-muricholic acid (αMCA), tauro-alpha-muricholic acid (T-α-MCA), tauro-beta-muricholic acid(T-β-MCA), taurochenodeoxycholic acid (TCDCA), taurocholic acid (TCA), taurodehydrocholic acid (THCA), taurodeoxycholic acid (TDCA), taurohyodeoxycholic acid (THDCA), taurolitocholic acid (TLCA), tauro-omega-muricholic acid (TωMCA) and tauroursodeoxycholic acid (TUDCA) and polar metabolites was done using alanine, citric acid, fumaric acid, glutamic acid, glycine, lactic acid, malic acid, 2-hydroxybutyric acid, 3-hydroxybutyric acid, linoleic acid, oleic acid, palmitic acid, stearic acid, cholesterol, fructose, glutamine, indole-3-propionic acid, isoleucine, leucine, proline, succinic acid, valine, asparagine, aspartic acid, arachidonic acid, glycerol-3-phosphate, lysine, methionine, ornithine, phenylalanine, serine and threonine. For other compounds detected, the results are given as normalized peak areas, after normalization with the closest eluting internal standard.
QC/QAQuality control was accomplished both for lipidomics, polar metabolites and PFAS analysis by including blanks, pure standard samples, extracted standard samples, pooled quality control samples, and standard reference plasma samples (NIST SRM 1950). The pooled sample was prepared by taking an aliqout (10 µl) of each extract, separately for lipidomic and polar metabolite methods, then pooling them, and aliquoting the pool into separate vials. In lipidomic and metabolomic analyses, lipids that had >30% RSD in the pooled QC samples (an equal aliquot of each sample pooled together) or that were present at high concentrations in the extracted blank samples (ratio between samples to blanks < 5) were excluded from the data analyses.
Statistical analysisData pre-processing and clusteringIn this study, all data analyses were conducted using the R statistical programming language (version 4.1.2) (https://www.r-project.org/). The exposure datasets were pre-processed by log2 transformation and scaling to zero mean and unit variance (auto-scaled). For contaminant exposure analyses, individual contaminant and cluster-level analyses were performed. To cluster the contaminant data, we utilized the ‘mclust’ R package (version 5.4.10) for model-based clustering, selecting the model type and the number of clusters based on the highest Bayesian Information Criterion (BIC). To better understand the impact of exposure, we also incorporated lipidomics and metabolomics data from our previous study [39], which included eight lipid clusters (LCs) and twelve polar metabolite clusters (PCs), along with their individual features (Supplementary Table 1).
Demographic data and covariatesIn terms of demographic data and covariates, the median age at the time of diagnosis for subjects who later developed autoimmune diseases was 15 years. We obtained information on birthweight, maternal age, gestational age, and BMI from the questionnaire. We have also data on lifestyle of the mothers and socioeconomic factors from both parents. We utilized birthweight and gestational age to calculate birthweight for gestational age (BWGA) Z-score, utilizing internationally validated infant growth charts developed by Fenton [46, 47].
Correlation and partial correlation analysisPairwise Spearman’s correlation between contaminants, lipid clusters (LCs), Polar metabolite clusters (PCs), and demographic variables (Z-score, Maternal age, BMI) was calculated and visualized using ‘corrplot’ R package (version 0.92). Two correlation plots were generated separately for control and cases. The correlation between variables visualised in the form of a matrix plot refers to positive and negative correlations and the strength of the association is referred to by the size of the dot or filled circles.
The Debiased Sparse Partial Correlation algorithm (DSPC) [48] was used to estimate partial correlation networks and visualized in the form of a chord diagram using ‘circlize’, R package (version 0.4.15) with edge ranges between ±0.14 to 1.0 and showing only correlations across contaminants, LCs, PCs, and demographic variables.
Univariate statistical analysisTo understand the impact of contaminant exposure levels on cord serum metabolome, the subjects were assigned to four quartiles based on the exposure levels. A two-way analysis of variance (ANOVA) test was performed followed by post-hoc Tukey’s test by using quartiles (Q1 to Q4) and subjects (cases and control) as factor variables. ANOVA test helps to identify any significant changes in the lipid or metabolite clusters and post-hoc Tukey’s test helps to identify the specific quartiles between which significant changes are observed.
Regression and classification analysisPredictive logistic ridge regression (LRR) was performed to investigate the impact of individual contaminants on the stratification of autoimmune cases and controls. We have adapted the L2 regularization strategy to avoid multicollinearity among highly correlated predictors. Regularized regression modelling was performed using the ‘glmnet’ package in R (version 4.1-4). The hyper-parameter λminimum was determined by 10-fold cross-validation using the ‘cv.glmnet’ function from ‘glmnet’. The models were adjusted for Z-score, Maternal age and BMI. The accuracy of prediction was determined by AUCs, where the mean AUC of the model was estimated by bootstrapping, by resampling the exposure dataset into training (80%) and testing (20%) 10,000 times. All LRR models with a threshold of AUC > 0.60 were considered. Downsampling was performed to address the class imbalance problem (cases, n = 62, controls, n > 62). The ‘caret’ package (version 4.1.3) was used for the partition of data and the best models (based on mean AUCs) were assessed using Receiver Operating Characterisitic (ROC) curves using the ‘ROCR’ package. Additionally, we have performed a stepwise recursive feature elimination scheme to identify the minimum number of predictors that are needed to maximize the outcome.
To investigate the effect of contaminant exposure on the cord blood metabolome, we employed linear regression with L2 regularization (LR), using individual contaminant concentrations as predictors and the concentrations of significantly altered cord blood lipid or polar metabolites (and their cluster) as the response variable. Note that we conducted linear regression with L2 regularization for cases and controls together. The hyper-parameter λminimum, which corresponds to the minimum cross-validation error, was selected through 10-fold cross-validation. We partitioned the data and performed resampling (10,000 iterations) as described earlier. The mean R square was used to estimate the accuracy of prediction and the significant impact of contaminant exposure on the cord blood metabolome.
Additionally, we determined the ranks of the predictors using LR and LRR modelling. For the LRR models, the ranks of the predictors were estimated based on the unit absolute differences in the odds ratio, while for the LR models, the ranks were based on the ridge coefficients normalized with the maximum value.
Pathway analysisPathway enrichment analysis comparing cases versus controls for Deoxynivalenol (DON) impact polar metabolites was performed using the MetaboAnalyst 5.0 web platform with the Functional Analysis (MS Peaks) module [49]. The input data for the pathway analysis consisted of complete high-resolution LC-MS spectral peak data obtained in negative ionization mode with a mass tolerance of 10 ppm. Linear regression analysis was performed to estimate the association between DON and polar metabolites while adjusting for Z-score, Maternal age, and BMI. The whole input peak list with FDR-corrected p-values and T-score was used for the pathway analysis. Overrepresented pathways were estimated against the background human scale metabolic model MNF (from MetaboAnalyst Mummichog package) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for Homo sapiens to determine the relative significance of the identified pathways [50]. The MetaboAnalyst 5.0 metabolomics pathway analysis (MetPA) tool [51] was used to calculate the Pathway Impact Scores [49, 52].
留言 (0)