
記住我
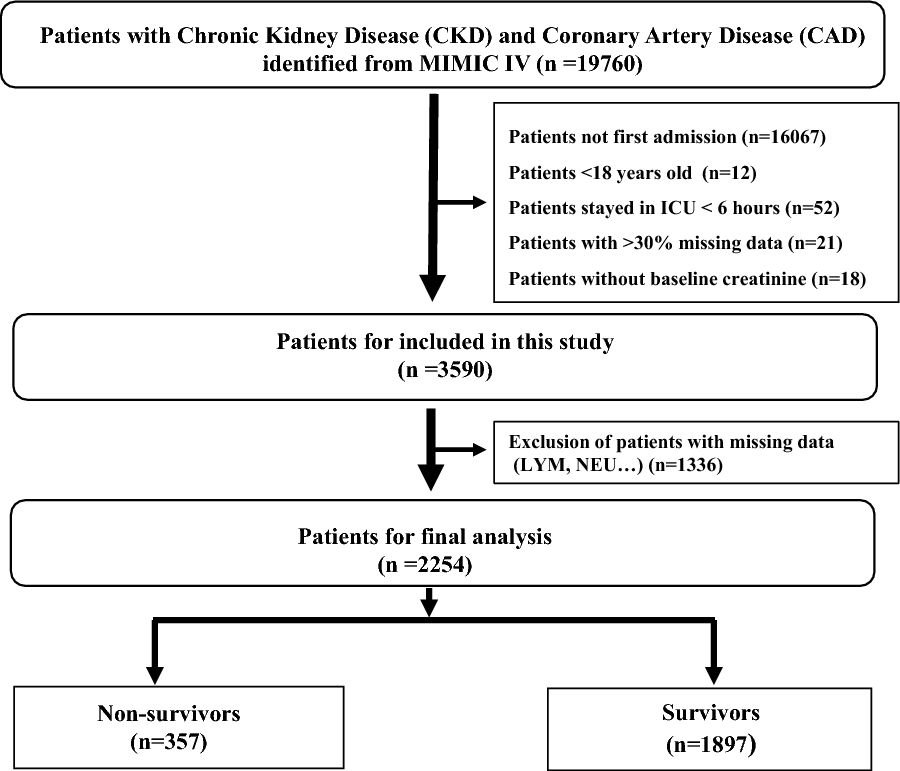




258 ERS-related genes were obtained and intersected with 344 OS-related prognostic genes; ERLIN2 and CDK5RAP3 were ultimately obtained as potentially ERS-related prognostic genes (Fig. 2A). Subsequently, ERLIN2 was verified to be lowly expressed in PCa and associated with a good prognosis in TCGA-PRAD, including OS and PFI (Fig. 2B), whereas CDK5RAP3 was highly expressed in PCa and associated with a bad prognosis (Fig. 2C). Moreover, ERLIN2 and CDK5RAP3 were negatively correlated, r = − 0.141, p = 0.002 (Fig. 2D). The relationship between basic clinical information and gene expression (ERLIN2 and CDK5RAP3) in the TCGA-PRAD cohort is shown in Additional file 1: Table S1. Association with clinic-pathologic features revealed that ERLIN2 was expressed significantly low in patients with high T stage (T3&4), M stage (M1), and partial remission (PR) (Fig. 2E–H), whereas CDK5RAP3 was expressed significantly high in patients with high T stage (T3&4), N stage (N1), and partial remission (PR) (Fig. 2E–H). Therefore, the present study tentatively suggests that ERLIN2 is a cancer suppressor gene and CDK5RAP3 is an oncogene. Interestingly, ERLIN2 and CDK5RAP3 were correlated with patients’ disease states of complete and partial remission, and this phenomenon implicated that they might influence the outcome of PCa treatment.
Fig. 2
Identification and characterization of prognostic ERS-related genes. A Venn diagram of ERS-related genes versus PCa prognostic OS-related genes; in TCGA-PRAD, B ERLIN2 differential expression, OS and PFI; C CDK5RAP3 differential expression, OS and PFI; D correlation of expression between ERLIN2 and CDK5RAP3; correlation between ERLIN2 and CDK5RAP3 with clinic-pathologic features, including E T stage, F N stage, G M stage, and H primary treatment outcome
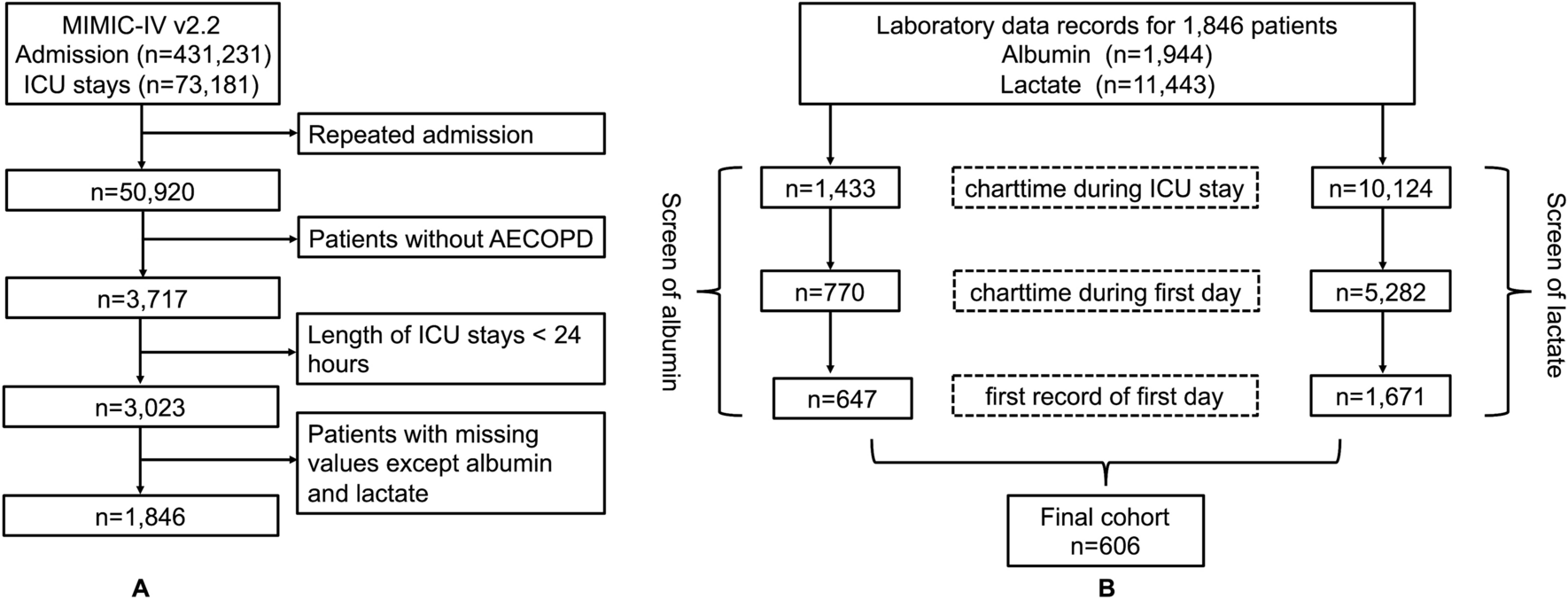
Due to the specificity of the PCa prognosis, this study first constructed the correlation between time-dependent ROC and univariate–multivariate Cox regression related to OS; However, its results were not statistically significant (Additional file 1: Fig. S1). Therefore, we continued to explore the nomogram with a focus on PFI. The 1-, 3-, and 5-year PFIs for AUCERLIN2 were 0.433, 0.431, and 0.392, respectively (Fig. 3A, B); and the 1-, 3-, and 5-year PFIs for AUCCDK5RAP3 were 0.647, 0.690, and 0.665, respectively (Fig. 3A, B). The inclusion of ERLIN2, CDK5RAP3 and clinical characteristics (TNM stage, primary therapy outcome) in the univariate–multivariate Cox regression, ultimately clarified that CDK5RAP3, T stage, and primary therapy outcome had independent prognostic value, but ERLIN2 multivariate Cox regression result of p = 0.06, was still included in the construction of the nomogram (Fig. 3C, D). Therefore, this study finally obtained the nomogram based on ERLIN2, CDK5RAP3, T stage, and primary therapy outcome, with C-index = 0.782, p < 2e−16 (Fig. 3E); the 3- and 5-year prognostic calibration curve was well fitted to the ideal curve (Fig. 3F).
Fig. 3
Construction of the PFI nomogram with the clinic-pathological features, ERLIN2 and CDK5RAP3. A PFI-associated time-dependent ROC for ERLIN2 and CDK5RAP3; B PFI-associated time-dependent AUC for ERLIN2 and CDK5RAP3; prognostic ERS-related genes combined with clinic-pathologic features in univariate (C) and multivariate (D) Cox regression analyses; E PFI nomogram; F 3-, 5-year prognostic calibration curves
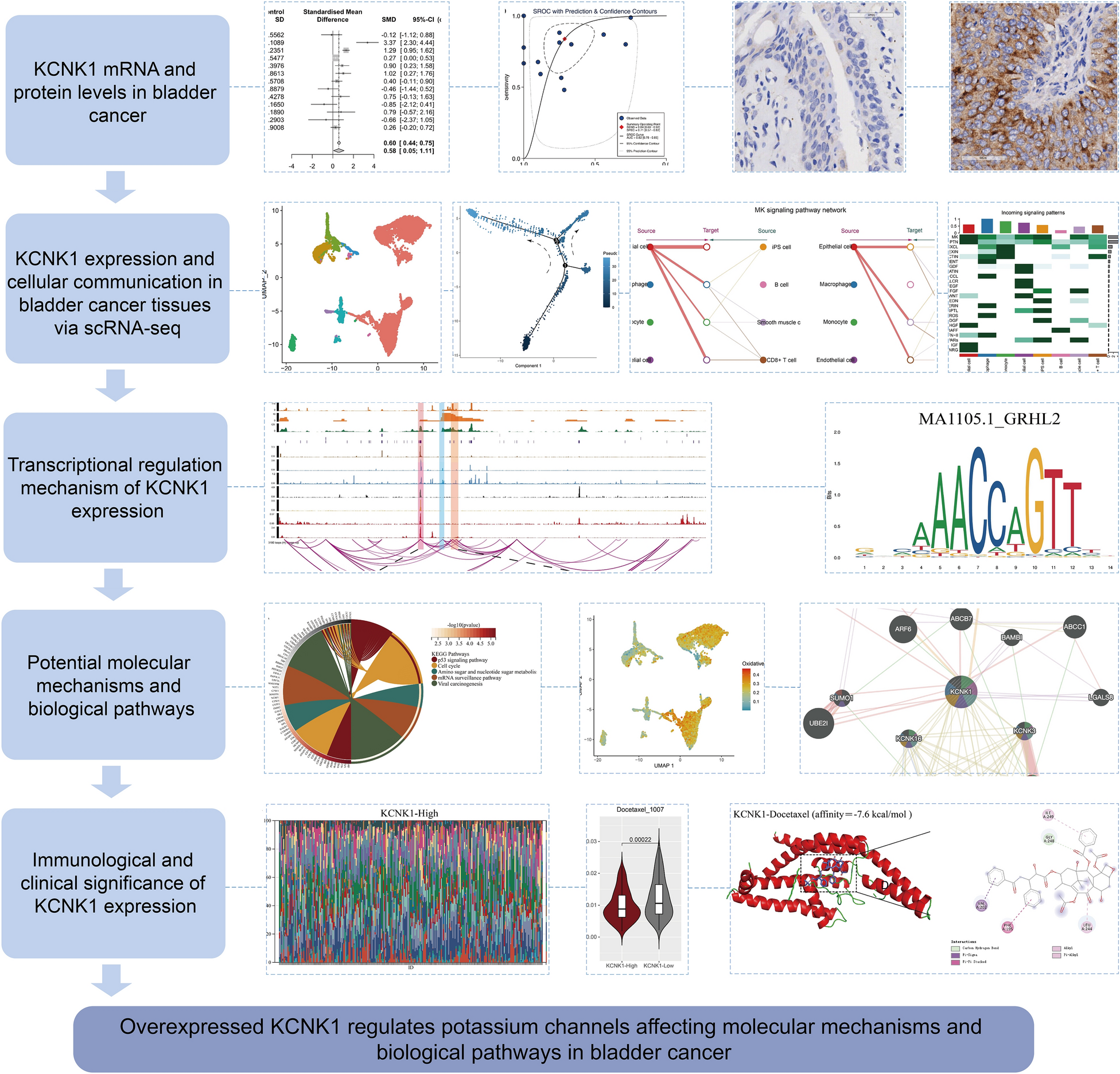
Characterization of ERLIN2 and CDK5RAP3 as key genes for immune infiltration and immunotherapy response in PCaThrough the xCELL algorithm, this study found that ERLIN2 and CDK5RAP3 were significantly associated with many tumor-immune cells (Fig. 4A, B). Given the immune microenvironment characterization, patients suffering from PCa having low ERLIN2 expression contained more M1 and M2 macrophages, whereas those with high CDK5RAP3 expression contained fewer M1 and M2 macrophages (Fig. 4A, B). Subsequently, further exploration of ERLIN2, CDK5RAP3, and immunotherapy correlation revealed that ERLIN2 was associated with immune checkpoint-related genes such as CD274, LAG3, PDCD1LG2, and SIGLEC15; whereas CDK5RAP3 was associated with CTLA4, LAG3, and PDCD1 (Fig. 4C). Furthermore, in the TCGA-PARD cohort, ERLIN2 expression was negatively correlated with TMB (p = 0.035, r = − 0.10) (Fig. 4D), whereas in the ICGA-PRAD cohort, CDK5RAP3 expression was negatively correlated with TMB (p = 6.44e−06, r = − 0.37) (Fig. 4E). In response to immune checkpoint blockade therapy, responsiveness to low ERLIN2 expression was higher in the TCGA-PARD cohort (Fig. 4F, G); responsiveness to low CDK5RAP3 expression was higher in the ICGA-PRAD cohort (Fig. 4F, G). These results all indicated that ERLIN2 and CDK5RAP3 affected the PCa TIM and had potential relevance to tumor immunotherapy.
Fig. 4
Single gene immunocorrelation analysis of ERLIN2 and CDK5RAP3. Analysis of ERLIN2 (A) and CDK5RAP3 (B) with immunocyte infiltration in the TCGA-PRAD cohort based on the xCELL algorithm; C correlation analysis of ERLIN2 and CDK5RAP3 with immune checkpoint-related genes; correlation analysis of ERLIN2 and CDK5RAP3 with tumor mutation burden (TMB) in TCGA-PRAD (D) and ICGA-PRAD (E); correlation of ERLIN2 and CDK5RAP3 with immune checkpoint blockade therapy (ICB) in TCGA-PRAD (F) and ICGA-PRAD (G)
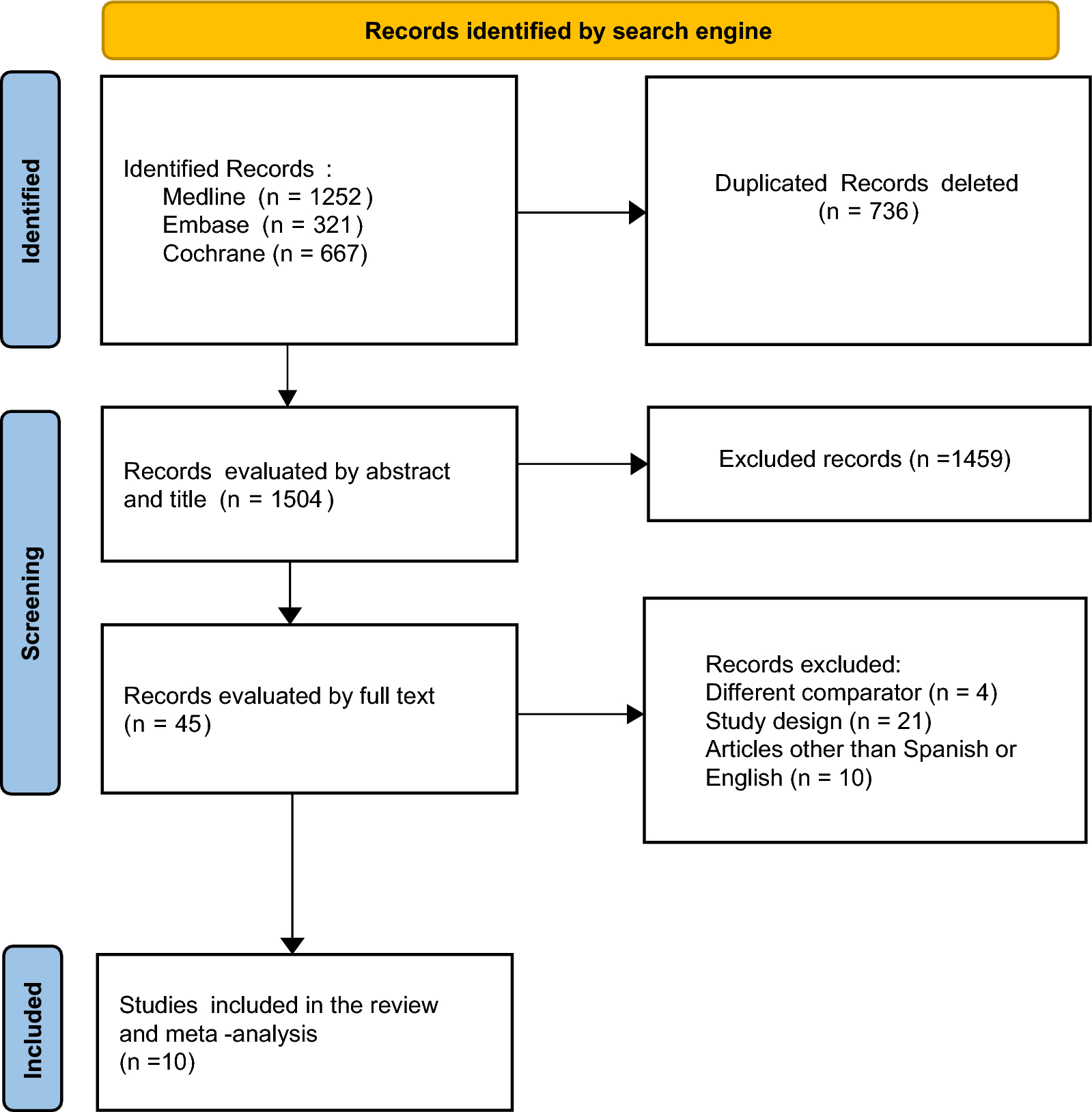
Construction of prognostic models and subgroups based on ERLIN2 and CDK5RAP3 in PCaThe prognostic model for PFI based on ERLIN2 and CDK5RAP3 was constructed using LASSO regression analysis with Riskscore = (− 0.1918) * ERLIN2 + (0.5254) * CDK5RAP3, lambda.min = 0.0022 (Fig. 5A, B). Analysis of ERLIN2 and CDK5RAP3 about the survival status of PCa showed that the number of survival statuses was lower in the high-risk group, with low ERLIN2and high CDK5RAP3 expressions (Fig. 5C). The analysis in the TCGA-PRAD cohort suggested a shorter PFI in the high-risk group (Fig. 5D), and the 1-, 3-, and 5-year PFI ROC were 0.652, 0.697, and 0.690, respectively (Fig. 5E). Based on the xCELL algorithm, the prognostic model risk score was analyzed for correlation with immune infiltration, suggesting that the model mainly showed a significant negative correlation with T cell CD4+ central memory and a positive correlation with common lymphoid progenitor (Additional file 1: Fig. S2). Moreover, this study also focused on the positive correlation between risk scores and macrophages (both M1 and M2) in this prognostic model (Additional file 1: Fig. S2).
Fig. 5
Construction of the PFI-related risk prognostic model about ERLIN2 and CDK5RAP3. A, B LASSO regression analysis; C risk score distribution, survival status, and expression of the two ERS-related genes in TCGA-PRAD; D Kaplan–Meier survival curves analyses; E time-dependent ROC curves
Because only two genes, ERLIN2 and CDK5RAP3, had the highest average concordance within the sample group based on K = 2, cluster analysis was performed to obtain two subgroups, with a total of 249 ERS high-risk and 249 ERS low-risk (Fig. 6A, B). Differential expression analysis of different subgroups suggested lower ERLIN2 expression and higher CDK5RAP3 expression in the ERS high-risk group relative to the ERS low-risk group (Fig. 6C). Combined with the results of OS and PFI, there was a decreasing trend in OS in the ERS high-risk group, but it was not statistically significant (Fig. 6D); however, PFI was significantly shorter in the ERS high-risk group (p = 0.01) (Fig. 6E).
Fig. 6
Identification of two cluster subgroups of ERS-related genes. A Cluster heatmap at K = 2; B area under the distribution curve; C differences in expression of ERLIN2 and CDK5RAP3 under different cluster subgroups; Kaplan–Meier curves for OS (D) and PFI (E) for different cluster subgroups
Identification of differentially expressed genes and pathways in different ERS-related subgroupsTo explore the molecular mechanisms underlying the prognostic differences caused by ERS-related subgroups, a total of 526 abnormally regulated genes were identified in the two subgroups, including 518 upregulated and 8 downregulated genes (FC ≥ 1.5, p < 0.05) (Fig. 7A). GO analysis revealed that upregulated genes were concentrated in endomembrane system, vesicle, golgi apparatus, and so on. (Fig. 7B); downregulated genes were found in nucleolus, RNA processing, bHLH transcription factor binding, and so on (Fig. 7C). KEGG analysis revealed that upregulated genes were concentrated in valine leucine and isoleucine degradation, arrhythmogenic right ventricular cardiomyopathy, proteoglycans in cancer, and metabolic pathways (Fig. 7D); KEGG analysis could not be performed for downregulated genes due to them being fewer in number. Moreover, Gene Set Enrichment Analysis (GSEA) indicated significant differences between ERS low-risk and ERS high-risk subgroups in multiple intracellular responses (Fig. 7E), metabolic responses (Fig. 7F), and oncogenic signaling pathways (Fig. 7G), which could be potential mechanisms leading to different prognosis. Interestingly, the study discovered that ANDROGEN RESPONSE and ESTROGEN RESPONSE had higher enrichment scores in the ERS high-risk subgroup (Fig. 7E), which may be an important factor influencing androgen deprivation therapy.
Fig. 7
Identification of differentially expressed genes and signaling pathways under different cluster subgroups. A Volcano and heat maps of differentially expressed genes (DEGs) profiles; B GO analysis of upregulated DEGs; C KEGG analysis of upregulated DEGs; D GO analysis of downregulated DEGs; E–G GSEA pathway enrichment analysis of DEGs
Through somatic mutation profiles, this study found significant differences in gene mutations between the ERS low-risk and ERS high-risk subgroups. The first 10 mutated genes in the ERS low-risk subgroup were SPOP, TP53, TTN, FOXA1, MUC16, SPTA1, SYNE1, CACNA1E, CSMD3, and KMT2D (Additional file 1: Fig. S3A); mutated genes in the ERS high-risk subgroup were TP53, TTN, SPOP, FOXA1, KMT2D, LRP1B, KMT2C, SYNE1, MUC16, and RYR1 (Additional file 1: Fig. S3B). Among them, the mutation rates of SPOP and MUC16 were significantly lower in the ERS high-risk than in the ERS low-risk subgroup.
Exploration of the immune relevance in different ERS-related gene subgroupsAs the above studies initially confirmed the correlation of ERLIN2 and CDK5RAP3 with the PCa-immune microenvironment and immunotherapy, the study delved into the correlation between prognostic subgroups based on ERLIN2-CDK5RAP3 with tumor immunity. By the xCELL and ESTIMATE algorithms, the ERS high-risk subgroup was identified to have higher ImmueScore, StromalScore, and ESTIMATE Score, and lower Tumor purity (Fig. 8A–C, E–H), and immune cell infiltration was explored under different subgroups according to the xCELL algorithm (Fig. 8D). IPS scores suggested higher MHC and AZ expression in ERS high-risk subgroup (Fig. 8I). Furthermore, as existing studies confirmed that both PCa and ERS are significantly correlated with macrophages, this study used the quantTIseq, xCELL, and CIBERSORT algorithms containing M1 and M2 macrophage subtypes, as well as discovered that quantTIseq suggested that the ERS high-risk subgroup had more M1 macrophages and fewer M2 macrophages (Fig. 8J), whereas xCELL suggested that the ERS high-risk subgroup had more both M1 and M2 macrophages (Fig. 8K), and there was no statistical significance for the CIBERSORT analysis (Additional file 1: Fig. S4C). Moreover, different ERS-related subgroups were confirmed to be significantly associated with macrophage by TIMER and EPIC algorithms (Additional file 1: Fig. S4A, B). Confusingly, there is not yet a clear homogeneity in macrophage expression for prognostic subgroups under different algorithms, which may be due to differences caused by different algorithmic folding or macrophage identification. Therefore, the correlation between ERS-related gene subgroups and macrophage expression needs to be further confirmed by experimental studies.
Fig. 8
Recognition of immune infiltration differences under different cluster subgroups. ImmueScore (A), StromalScore (B) and MicroenvironmentScore (C) based on the xCELL algorithm; D immunocyte infiltration stack plot for different cluster subgroups under the xCELL algorithm; ImmueScore (E), StromalScore (F), ESTIMATE score (G) and tumorpurity (H) based on the ESTIMATE algorithm; I IPS scores for different cluster subgroups; differential expression of M1 and M2 macrophages under quanTIseq (J) and xCELL algorithm (K) with different cluster subgroups
Subsequently, this study thoroughly explored the correlation of different subgroups with immune molecules, including immunostimulators, chemokines, receptors, immunoinhibitors, and MHC molecules. Multiple immunostimulators were expressed significantly low in the ERS high-risk subgroup (p < 0.001), including CD276, CXCL12, ENTPD1, IL6R, NT5E, PVR, RAET1E, TNFRSF18, TNFRSF25, TNFRSF4, TNFSF13, TNFSF15, and TNFSF4 (Fig. 9A). Additionally, the immunoinhibitor LAG3 was significantly highly expressed in the ERS high-risk subgroup (p < 0.001), but CD160, CD274, KDR, PDCD1LG2, and TGFBR1 were expressed significantly less (Fig. 9D). The chemokine CCL17 was significantly highly expressed in the ERS high-risk subgroup, whereas CCL28, CXCL12, and CXCL16 were expressed significantly less (Fig. 9B). The ERS high-risk subgroup showed significantly low expression of the receptors CCR2, CXCR1, CXCR2, XCR1, and CX3CR1 (Fig. 9C). Finally, for MHC molecules, only B2M showed significantly low expression in the ERS high-risk subgroup (Fig. 9E).
Fig. 9
Identification of immune-related molecules expression under different cluster subgroups. A Immunostimulators; B chemokines; C receptors; D immunoinhibitors; E MHC molecules
Verification of ERLIN2 and CDK5RAP3 expression in PCa by cell phenotype and immunohistochemistryThe relative wound healing rate of cells was found to be significantly higher after siERLIN2 and significantly lower after siCDK5RAP3 by cell scratch assay (Fig. 10A, B). Then, CCK8 experiments suggested accelerated cell proliferation after siERLIN2, whereas cell proliferation was significantly weakened after siCDK5RAP3 (Fig. 10G–J). In addition, the number of cell clones increased after siERLIN2, while the number of cell clones decreased after siCDK5RAP3, compared with the ncRNA group (Fig. 10C, D). Moreover, the cell migration ability was elevated after siERLIN2, whereas it was decreased after siERLIN2 (Fig. 10E, F). Collectively, the above studies further confirmed ERLIN2 as an anti-oncogene and CDK5RAP3 as a pro-oncogene in PCa.
Fig. 10
ERLIN2 and CDK5RAP3 were proved to promote prostate cancer progression in vivo. Scratch tests in DU145 (A) and LNCaP (B); clone formation assay in DU145 (C) and LNCaP (D); migration assay in DU145 (E) and LNCaP (F); ERLIN2 (G) and CDK5RAP3 (H), CCK8 assay in DU145; ERLIN2 (I) and CDK5RAP3 (J) CCK8 assay in LNCaP; left, wound healing and migration assay, scale bar, 100 μm
Through the HPA database, immunohistochemical images and patient information under different antibody identifications were obtained (Additional file 1: Table S2). Antibodies HPA002025 (Fig. 11A) and CAB014894 (Fig. 11B) directed against ERLIN2 suggested that ERLIN2 had low expression in PCa tissues; antibodies HPA022141 (Fig. 11C), HPA022882 (Fig. 11D), and HPA027883 (Fig. 11E) directed against CDK5RAP3 suggested that CDK5RAP3 was highly expressed in PCa tissues. The above results were consistent with the results of the previous bioinformatic analyses.
Fig. 11
Immunohistochemistry of ERLIN2 and CDK5RAP3 in the HPA database. A, B ERLIN2; C–E CDK5RAP3
留言 (0)