
記住我


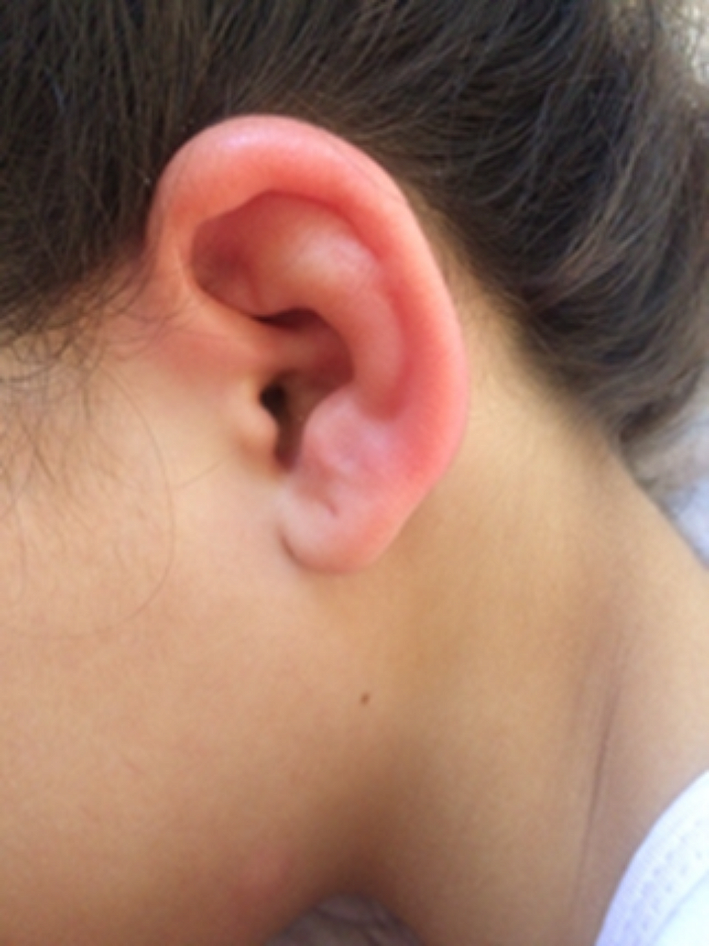



The frequency of ocular involvement is approximately 20% in patients with RP at the disease presentation, reaching up to 65% of patients during follow-up. Episcleritis or scleritis are the most frequent manifestations of the disease and in patients with long-term disease, reactive lymphoid hyperplasia results in a salmon-colored mass in the conjunctiva. Scleritis may be the first manifestation whose study leads to the diagnosis of RP. Scleritis associated with RP is more often bilateral, necrotizing, recurrent and associated with decrease of vision than scleritis associated with other systemic immune-mediated diseases [14, 15]. Recurrent bouts of ocular inflammation can cause thinning of the sclera, allowing a dark or even bluish color of the choroid to be seen through the thinned sclera. Keratoconjunctivitis sicca, keratitis, corneal perforation, iritis, retinopathy and optic neuritis can also occur in RP and may result in blindness. External inflammation of the eyeball can present as an orbital pseudotumor, eyelid edema or extraorbital muscle paralysis. (Fig. 4) [14].
Fig. 4
Episcleritis in an adolescent with RP. (Author’s personal archive)
Musculoskeletal manifestationsArthralgias and inflammatory arthritis may occur during the course of the disease in 70 to 80% of patients with RP. Serum-negative arthritis is typically asymmetrical, has a migratory pattern and frequently involves the sternoclavicular, costochondral, and manubriosternal joints. Intermittent exacerbations of arthritis are usually observed. Some cases show reduced joint space and osteopenia on X-rays. In severe cases, the clavicles and ribs may dislocate due to cartilage lysis resulting in an unstable chest [12–13].
Mucocutaneous manifestationsMucocutaneous changes are present in half of patients with RP. The skin is involved in 20 to 30% of patients with the primary form of RP and in up to 90% of patients with RP associated with myelodysplastic syndrome (MDS). Oral ulcers are the most common RP mucocutaneous manifestations. Oral and genital ulcers associated with cartilaginous inflammation known as MAGIC syndrome (Mouth And Genital ulcers with Inflamed Cartilage) represent an overlap between Behcet’s syndrome and RP. Other manifestations include erythema nodosum, purpuric lesions, livedo reticularis, urticaria, angioedema, erythema multiforme, and panniculitis. Histological examination of skin biopsies reveals leukocytoclastic vasculitis, thrombosis of cutaneous vessels, septal panniculitis, and neutrophilic dermatosis [13]. The presence of mucocutaneous lesions in a patient with RP should lead to the suspicion of underlying myelodysplasia and VEXAS syndrome, which is an autoinflammatory disease associated with a somatic mutation of the UBA1 gene [16].
Hematological manifestationsHematological manifestations of primary RP are infrequent [12–13]. In the presence of myelodysplastic syndromes in association with polychondritis, especially in men between 60 and 70 years of age, with hematologic abnormalities such as macrocytic anemia, cytopenias, multiple myeloma or monoclonal gammopathy of undetermined significance (MGUS), genotyping of the UBA1 gene is mandatory to diagnose the recently described VEXAS syndrome. In this condition, vacuoles are observed in myeloid precursor cells in the bone marrow in 100% of reported cases. Venous thrombosis, arterial thrombosis and macrophage activation syndrome may also occur [16].
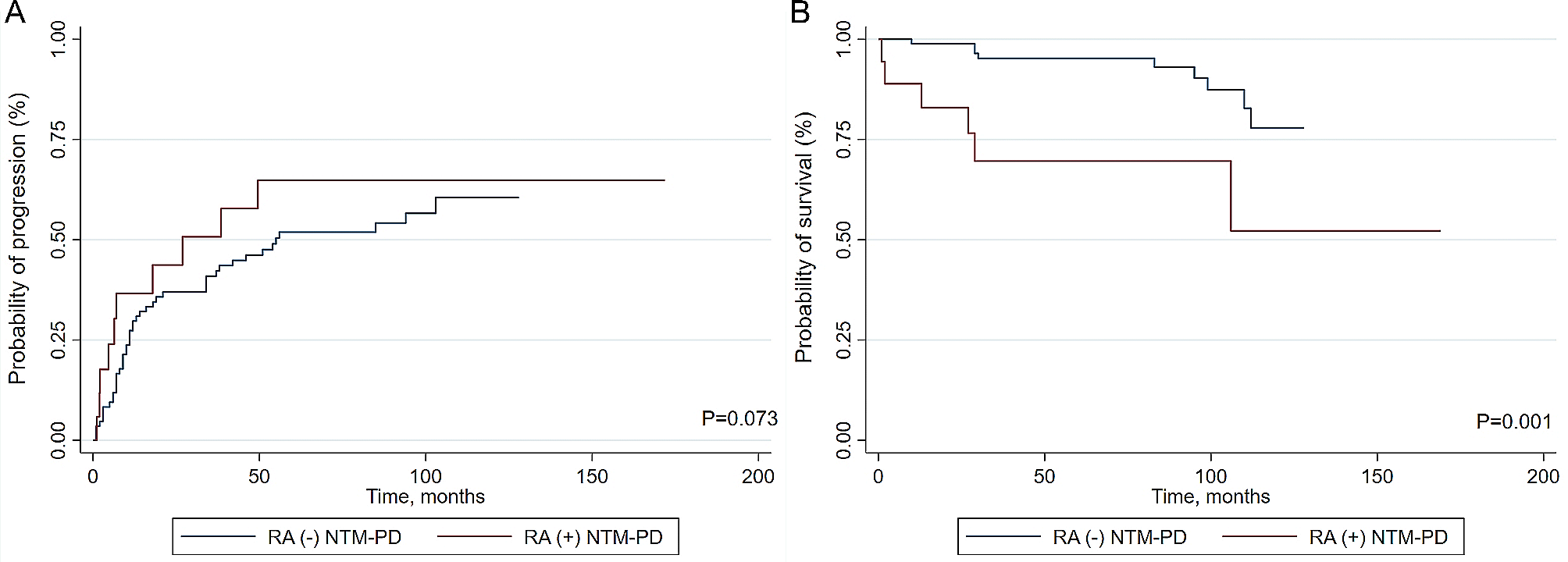
Cardiovascular manifestationsThe cardiovascular manifestations of RP typically occur in patients with long-standing disease, even when these patients are being treated with immunosuppressive therapy. Aortic insufficiency is the most common cardiovascular complication of RP occurring in 4 to 10% of patients. Mitral regurgitation occurs in 2% of patients with RP. Other manifestations include aortitis, abdominal and thoracic aortic aneurysms with aneurysmal dilation of the aortic arch, conduction abnormalities, pericarditis, as well as arterial thrombosis in case of associated antiphospholipid antibodies [17] (Fig. 5).
Fig. 5
Chest angiotomography showing a thoracic aortic aneurysm in an adolescent with RP. (Author’s personal archive)
Neurological manifestationsNeurological involvement occurs in less than 3% of patients with RP but can present acutely or subacutely. The cranial nerves are frequently affected by RP, especially the second, sixth, seventh and eighth cranial nerves. The involvement of the second cranial nerve manifests as optic neuritis, while the paralysis of the lateral rectus is a feature of the sixth pair involvement. Paralysis and weakness of the facial nerve indicate the involvement of the seventh and vestibular dysfunction is typically observed when the eighth cranial nerve is affected. Furthermore, headache, seizures, cerebellar dysfunction with ataxia, mental confusion, cerebral aneurysm and aseptic meningitis have also been described in patients with RP [13].
Renal manifestationsThe kidney is an organ rarely affected in RP when it is not associated with other rheumatological diseases. Renal involvement is a sign of poor prognosis in RP. When renal dysfunction occurs, a possible association with systemic lupus erythematosus (SLE) or other systemic diseases should be investigated [13].
DiagnosisThe diagnosis of RP may be delayed in patients presenting only non-specific symptoms such as fever, weight loss, and fatigue, in the absence of signs of cartilage inflammation.
The diagnosis of RP is primarily based on a combination of clinical features, radiographic findings, and/or biopsy of a cartilaginous site [18]. There is no specific diagnostic test for RP.
Several clinical diagnostic criteria were developed for this disease [18,19,20]. Hence, tissue biopsy is not always necessary if there is enough clinical evidence.
McAdam et al. introduced the clinical criteria for RP in 1976 [19].
These clinical criteria were expanded upon by Damiani et al. in 1979 [20].
Michet et al. modified the criteria in 1986 [21] (Table 1).
Table 1 Box 1: RP diagnostic criteria [19,20,21]Laboratory tests and imagingThere is no specific laboratory test for diagnosing RP. Acute phase reactants may be normal even during disease exacerbation, and abnormal results are nonspecific. Cartilage-specific antibodies are not available in clinical practice and the detection of other autoantibodies (i.e., ANA, RF, ANCA, etc.) is useful for investigating diseases that may be associated with RP. Anti-type II collagen antibody tests are not routinely available, and when these tests are done, not all RP patients present positive results. There are no surrogate markers for ongoing cartilage damage [13].
Pulmonary function tests and bronchoscopy should be indicated on an individual basis. Pulmonary function testing is done to assess airway trapping and lung volumes. Positron Emission tomography with computed tomography (PET-CT) is a new diagnostic modality that seems to be helpful in early disease recognition and in providing a site for targeted biopsy.
Imaging tests such as X-rays, CT scans, and magnetic resonance imaging (MRI) can help evaluate the disease extent and highlight occult airway involvement. An initial screening should be performed in all patients recently diagnosed with RP. PET-CT has proven to be a potentially powerful tool for early diagnosis, especially in patients without organ involvement that is easily accessible for biopsy. This imaging modality also improves the assessment of the extent and disease activity during treatment.
Differential diagnosisChondritis or inflammation of the outer ear can occur due to trauma, such as those that occur in jiu jitsu fighters, or due to infection. In these cases, the chondritis is generally unilateral and also affects the ear lobe. Bilateral auricular chondritis, recurrent and spontaneously resolving, strongly suggests the diagnosis of RP. Other systemic diseases, such as granulomatosis with polyangiitis, rheumatoid arthritis, syphilis, reactive arthritis due to ocular involvement and sarcoidosis, may share some clinical characteristics with RP [13].
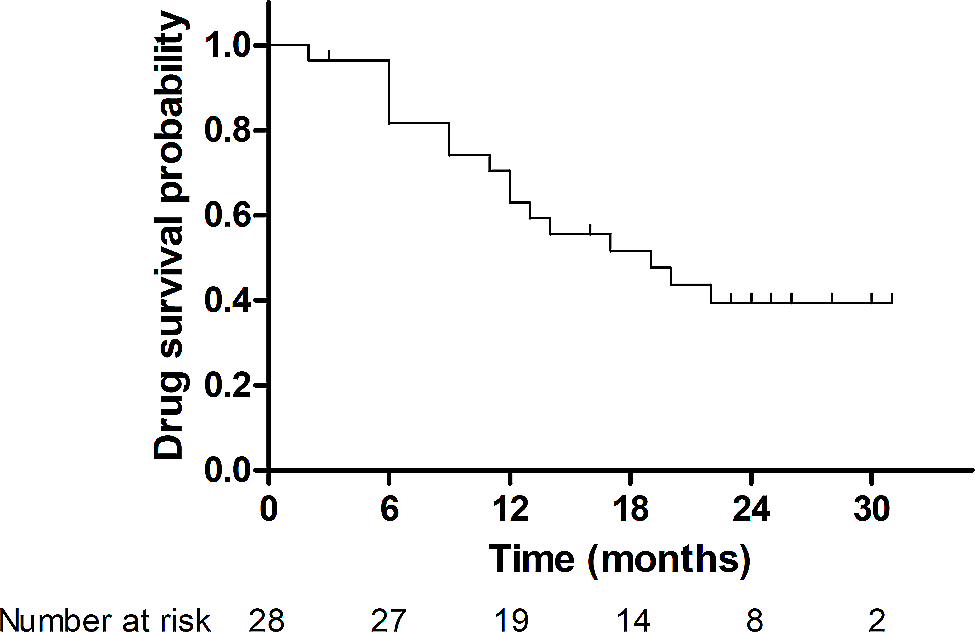
TreatmentBecause of its rarity, randomized, placebo-controlled studies have not been conducted in patients with RP. In mild cases of nasal or ear chondritis, non-steroidal anti-inflammatory drugs can be used. If a complete response is not achieved, corticosteroids such as prednisone or equivalent can be used at a dose of 40 to 60 mg/day. In relapsing cases or when other organs are involved, immunosuppressants such as methotrexate, azathioprine, cyclophosphamide, and cyclosporine may be prescribed. Involvement of other organs or systems may require specific interventions (e.g., surgical approach for aneurysm correction, abnormalities in the cardiac conduction system, tracheal stenosis, etc.) [22,23,24,25].
The use of biologic agents was evaluated in a multicentre retrospective cohort study in France [22]. This study aimed to evaluate the effectiveness and safety of biologic therapy in RP [22]. Forty-one patients exposed to 105 biologics for 1 year were analyzed. The biologics included TNF inhibitors, tocilizumab, anakinra, rituximab, and abatacept. The overall response rate during the first six months of exposure was 63%, but during this period they were used in combination with corticosteroids. The complete response rate to therapy was 19%. There was a trend toward a higher response rate in patients concomitantly exposed to nonbiologic disease-modifying antirheumatic drugs [22,23,24,25].
For patients with severe disease manifestations, other agents are used including JAK inhibitors [26].
VEXAS syndromeWithin the spectrum of polychondritis, a new syndrome was recently identified in which patients develop inflammatory and hematological symptoms. This new syndrome was named VEXAS syndrome and it is caused by a somatic mutation in the UBA1 gene in blood cells and appears late in life (i.e., above the age of 50 years). It is an X-linked disease and has therefore been primarily observed in men [27, 28].
VEXAS is often diagnosed as relapsing polychondritis refractory to treatment and it can remain misdiagnosed for many years. The detection of UBA1 gene mutations in patients with RP ranges from 7.6 to 72.7% [33].
VEXAS is an acronym for Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic [27].
V - vacuoles are often seen in cells identified in bone marrow aspirates from patients with VEXAS syndrome.
E - E1 ubiquitin-activating enzyme is encoded by the UBA1 gene, which is mutated in patients.
X - the UBA1 gene is located on the X chromosome.
A - patients have autoinflammation.
S - the mutations are somatic, meaning they are acquired at some point in life and are not inherited.
PathogenesisVEXAS is an auto-inflammatory disease, identified by a somatic mutation in a gene that encodes E1 enzymes, located on the X chromosome. All patients diagnosed with this syndrome presented cytoplasmic vacuoles, predominantly located in promyelocytes, myelocytes, erythroid precursors and blasts in the bone marrow [27].
The first case was described in 2020, in a man with an inflammatory disease associated with myelodysplasia. In genetic analysis, a mutation in the UBA1 gene was identified.
The UBA1 gene is located on the X chromosome and encodes the E1 enzyme, which is the main enzyme that initiates ubiquitination. Ubiquitin is a protein found in eukaryotic cells comprising 76 amino acids, this protein plays an important role in protein regulation, marking unwanted proteins so that they are degraded by a multiprotein complex called the proteasome.
Monocytes carrying UBA1 variants (mutants) showed decreased ubiquitination and activated innate immune pathways, causing systemic inflammation [27,28,29].
Mutation in the UBA1 gene occurs in somatic cells, producing systemic inflammation. In this sense, VEXAS has been called “haemato-inflammatory disease”. This condition manifests as a premalignant condition as it results in myeloproliferation, myelodysplasia, lymphoproliferation, or cellular malignant transformation potential [29].
Somatic mutations in UBA1 originate in bone marrow stem cells, affecting methionine-41 in the UBA1 gene, and are restricted to the myeloid cell lineage in peripheral blood. This change gives rise to a complex inflammatory syndrome that manifests itself in adulthood [30].
Sequencing of isolated cell populations revealed that UBA1 variants were found in more than half of progenitor and myeloid lineage cells, but were absent in T cells, B cells, and fibroblasts.
Clinical featuresIn the initial report, 25 patients were diagnosed with VEXAS syndrome based on the confirmation of somatic mutations in codon 41 of UBA1. The median age at disease onset was 64 years, and all patients were male. Auricular and/or nasal chondritis was one of the most common organ manifestations, with 15 (60%) patients meeting the classification criteria for RP. According to other cohort studies, the incidence of chondritis is 36–50% [11].
The typical presentation of VEXAS includes fever and systemic features resulting from inflammation, that involves the skin (leukocytoclastic vasculitis and neutrophilic dermatoses), lungs (alveolitis or exudative serositis), cartilage (polychondritis), and blood vessels [12, 27,28,29].
In general, in its presentation, macrocytosis is identified (mean corpuscular volume > 100 fl.), with progression to myelosuppression characterized by anemia, thrombocytopenia, and lymphopenia. Hematologic disorders are prevalent in VEXAS syndrome, including myelodysplastic syndrome (MDS), multiple myeloma, and monoclonal gammopathy of undetermined significance. Thromboembolic events are also common. The reported incidence of venous thromboembolism (i.e., 36.4%) is much higher than that of arterial thrombosis (1.6%) [42].
VEXAS has a characteristic that differentiates it from most monogenic inflammatory diseases: the identified mutation is present only in cells of somatic origin and not in germ cells [28].
Although the somatic mutations in UBA1 confirmed the diagnosis of all patients, it is not a widely available test, much less routinely applied.
A fact that draws attention is that patients with VEXAS syndrome meet the criteria for several other conditions, such as RP, polyarteritis nodosa, SLE, and Multiple Myeloma. Chondritis of the ears and nose is a manifestation of VEXAS and up to 8 to 14% of patients with RP may truly have VEXAS as an underlying diagnosis. The clinical spectrum of presentation is still expanding as it is a disease described very recently [16].
Patients with VEXAS may develop a wide range of inflammatory symptoms affecting multiple organs [29, 32–33]. A summary of clinical manifestations is presented in Table 2.
Table 2 Summary of clinical characteristics of VEXAS syndromeCutaneous manifestations are present in 90% of the patients whose cases have been described.
Acute phase reactant levels are increased in most patients with RP.
On many occasions, patients with VEXAS have an associated disease, including RP, polyarteritis nodosa, Sweet’s syndrome and myelodysplastic syndrome.
The presence of cytoplasmic vacuolation, characteristic of the disease, constitutes a useful marker for its identification and exclusion from differential diagnoses.
Currently, the diagnosis of this syndrome depends solely on the presence of UBA1 mutations confirmed by Sanger sequencing or next-generation sequencing (NGS), including whole-genome sequencing and whole-exome sequencing, that may favorably serve in the faster and more accurate diagnosis [32, 33].
TreatmentThe optimal treatment of VEXAS is still unknown and patients present a variable response. As it is a relatively new disease, effective therapies still need to be further investigated. Symptoms tend to be refractory, and high-dose corticosteroid therapy appears only temporarily effective and with considerable toxicity.
Immunosuppressants (e.g., Cyclophosphamide, Methotrexate, Mycophenolate mofetil and Azathioprine) and biologic agents (e.g., anti-IL6 receptor tocilizumab) [34, 37], anti-IL-17 (e.g., secukinumab)[35], abatacept [36], anti-IL1 inhibitors [37] (e.g., anakinra, canakinumab), TNF inhibitors [37] (e.g., infliximab and adalimumab), anti-CD20 (e.g., rituximab), and anti-IL-12/IL-23 (e.g., ustekinumab) therapies have been administered to patients with VEXAS syndrome, as well as inhibitors of JAK kinases [38] (e.g., ruxolitinib, tofacitinib and baricitinib), whose efficacy and safety still need to be studied. Given that multiple cytokines are involved in the disease mechanism of VEXAS syndrome, the use of JAK inhibitors seems better therapeutic strategy rather than single cytokine blockade.
One study evaluated the use of azacytidine [39] (a DNA hypomethylating agent), and observed a reduction in inflammatory symptoms for approximately 22 months, but without effectiveness in myelodysplasia [39]. Considering the high prevalence of MDS in VEXAS syndrome, azacytidine could be a good candidate for this syndrome.
If the above-mentioned therapies fail, allogeneic hematopoietic stem cell transplantation (ASCT) may be the last treatment option for VEXAS syndrome and a series of cases showing successful treatment with ASCT has been accumulated [40,41,42].
Patients still have high mortality due to the high refractoriness rate.
At this time, management must be individualized, based on each patient’s tolerability and response, and on the combined experience of the rheumatologist and hematologist following the patient.
留言 (0)