
記住我





FD is the second most common sphingolipidosis after GD. It is a pan-ethnic X-linked disease with an estimated birth incidence of 1 per 117,000 in the general population, and 1 per 40,000 male live births [8, 36, 37]. FD is caused by deficient or absent lysosomal α-galactosidase A (α-GAL) activity. This enzyme is encoded by the GLA gene located on the long arm of the X chromosome. More than 1000 GLA pathogenic or likely pathogenic variants have been described to date [8, 38].
α-GAL catalyzes the removal of terminal galactose groups from substrates such as globotriaosylceramide (Gb3) and glycoproteins [39]. Thus, the main metabolic consequence of α-GAL deficiency is the accumulation of Gb3 and its deacylated form globotriaosylsphingosine (lyso-Gb3) in multiple cells, especially in the vascular endothelium, vascular smooth muscle cells, cardiomyocytes, podocytes, autonomic ganglia, and conduction fibers [38,39,40,41].
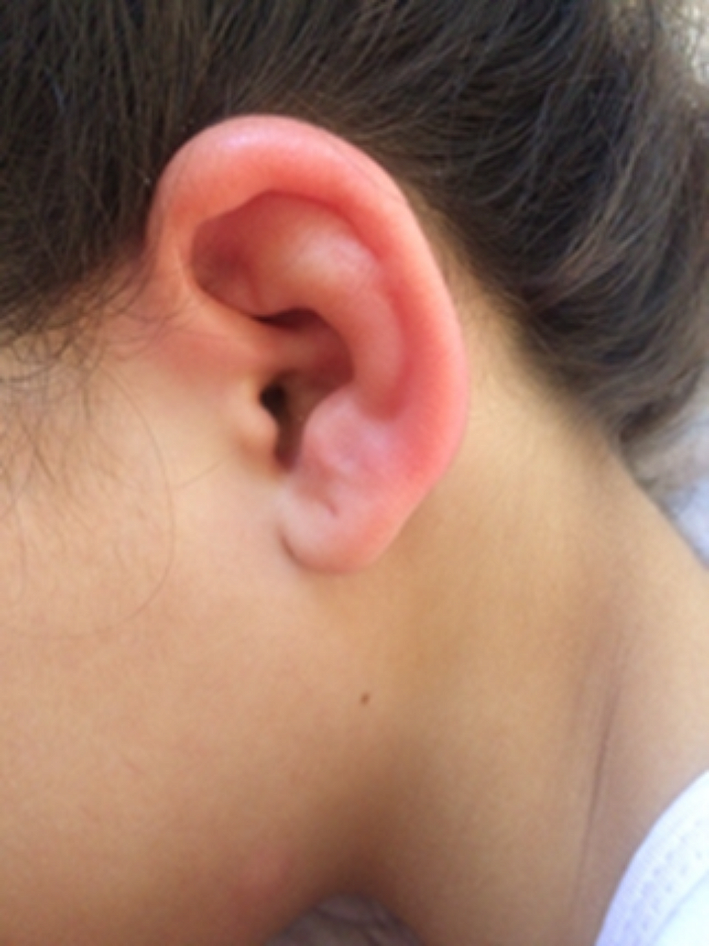
Manifestations of classic FD begin in childhood or adolescence. Classic FD is observed in hemizygous males, who have minimal or no enzymatic activity of α-GAL [42, 43]. Patients may present with a variety of manifestations, including neuropathic pain in the extremities (acroparesthesias); hypohidrosis; heat, cold, and exercise intolerance; gastrointestinal symptoms, such as recurrent abdominal pain, nausea, intermittent diarrhea, or constipation; unexplained fever; angiokeratomas, characterized as nonblanching red to bluish-black papules, most commonly on the trunk, groin, and periumbilical areas (Fig. 3); and corneal opacities (cornea verticillata) seen on slit lamp examination [42,43,44]. In adulthood, progressive organ failure becomes more evident with the following involvements: cardiac (left ventricular hypertrophy, arrhythmias, coronary artery disease, and heart failure), cerebrovascular (transient ischemic attacks and strokes), and renal (proteinuria and deterioration of kidney function) [42, 43].
Fig. 3
Multiple reddish-purple angiokeratomas in the lower abdomen of a patient with Fabry disease. Courtesy of Dr. Nilton Salles Rosa Neto
Female patients with FD are most frequently heterozygous for pathogenic variants in the GLA gene. They have a broad phenotypic presentation that ranges from no manifestation to a severe phenotype similar to classic FD, usually observed in hemizygous males [43, 45]. One explanation for this wide phenotypic variation in females is the skewed inactivation of one X chromosome in each cell during embryogenesis. Thus, clinical manifestations depend on the proportion of cells with the X chromosome that express the mutated GLA gene [46]. There are also male and female patients with variants of late-onset FD, who tend to have disease predominantly affecting the heart or the kidney [47,48,49].
Relevance for rheumatologistsFD is a relatively rare disease, with a wide variety of nonspecific manifestations. The lack of recognition of FD manifestations by the medical community may explain, at least in part, the significant delay in diagnosis, reported in the literature as a median time of 16 to 18 years [50,51,52]. Rheumatology may be one of the specialties sought by patients with FD during their journey to diagnosis. In fact, many FD manifestations may mimic rheumatic conditions, and it is not rare for these patients to be misdiagnosed and incorrectly treated [50,51,52].
A retrospective study that analyzed the medical records of 107 adult patients with FD found that 26.2% of them received at least one incorrect diagnosis of rheumatic disease. The authors argued that arthralgia, unexplained fever, Raynaud’s phenomenon, and episodes of elevated inflammatory markers were possible reasons for the incorrect diagnosis of a rheumatic condition [51]. Another study conducted with 37 Brazilian patients with definite FD reported that diagnostic errors occurred in 64.8% of the sample, and the majority of the incorrect diagnoses were musculoskeletal/rheumatic conditions, such as rheumatic fever, “unspecified rheumatism”, growing pains, and fibromyalgia [52].
In children and adolescents, rheumatologists should be alert to acroparesthesias, hypohidrosis, angiokeratomas, intolerance to exercise, heat or cold, and fever attacks with painful extremities without synovial inflammation as clues for early diagnosis of FD. A detailed family history is another extremely helpful point for diagnosing FD [53,54,55,56].
FD should be included in the differential diagnosis of recurrent fever of unknown origin, especially when associated with one or more of the characteristics mentioned above [57, 58]. In rheumatology practice, at first glance, fibromyalgia can also be confused with FD due to a history of multiple symptoms, including chronic pain and gastrointestinal complaints, which could be interpreted as irritable bowel syndrome in the context of central sensitization. However, a thorough history and physical examination may reveal atypical features not explained by fibromyalgia alone, although fibromyalgia syndrome may be present as a comorbidity in patients with FD [52, 59, 60].
DiagnosisThe diagnosis of FD is preferably made by the determination of α-GAL activity and/or genetic testing. In males with compatible FD phenotype, a very low leukocyte α-GAL activity (<5% of mean normal activity) is sufficient to establish the diagnosis. In females, genetic testing with detection of disease-causing variant in the GLA gene is required for the diagnosis of FD, since heterozygous females have variable α-GAL activity levels, ranging from normal to very low [42, 61, 62].
Mutational analysis of the GLA gene is useful regardless of sex, because it facilitates genetic counseling and allows identification of the amenability of gene variants to chaperone therapy [61, 63]. A small number of pathogenic variants, particularly in the case of duplications/deletions and deep intronic variants, may escape detection by routine analysis and can only be identified by a more sophisticated assessment of the GLA gene [61].
TreatmentTreatment for FD should be considered for all male patients, symptomatic or asymptomatic, with a pathogenic variant known to be associated with classical disease, regardless of the age of presentation. For females, treatment should be considered for symptomatic patients and/or those with evidence of major organ involvement. Asymptomatic females without laboratory, histological, or imaging evidence of renal, cardiac, or central nervous system involvement may not receive specific treatment but should be monitored regularly. Treatment should also be considered for male and female patients with later-onset variants and single-organ disease, provided that the abnormalities are attributable to FD [62].
FD-specific treatment consists of ERT, which involves biweekly intravenous infusions of recombinant α-GAL (agalsidase alfa, agalsidase beta, or pegunigalsidase alfa), or oral chaperone therapy (migalastat hydrochloride) administered every other day [64,65,66]. Treatment with ERT is recommended for both adults and children, but the age at which it is authorized varies according to the product and the approval specific to each country. Chaperone therapy stabilizes some misfolded forms of α-GAL, which facilitates their trafficking to lysosomes, prolongs their half-life and enhances their catalytic activity [66, 67]. Treatment with migalastat is intended for patients who are 12 years of age or older, have amenable GLA gene variants, and have an estimated glomerular filtration rate (eGFR) greater than 30 mL/min/1.73 m2. Amenability refers to the ability of the α-GAL encoded by amenable variants to respond to chaperone therapy in vitro with an increase in its activity. However, this does not necessarily imply a response in vivo, which should be monitored at the discretion of the attending physician [66, 67].
The therapeutic goals of specific treatments for FD are to improve quality of life and exercise tolerance; prevent the development or stabilize the progression of left ventricular hypertrophy, myocardial fibrosis, arrhythmias, and heart failure; reduce proteinuria and prevent the development or stabilize the progression of eGFR decline; decrease the risk of ischemic cerebral events; reduce the intensity of neuropathic pain and/or frequency of pain crises; and reduce gastrointestinal symptoms [42, 68].
Nonspecific treatments also play an important role in the management of FD and can include, but are not limited to, optimal blood pressure control; insertion of an implantable cardioverter defibrillator in patients with sustained ventricular arrhythmia or other high-risk situations; pacemaker implantation in patients with atrioventricular block; oral anticoagulation for atrial fibrillation; renal replacement therapy or kidney transplantation for end-stage kidney disease; and symptomatic treatment of chronic neuropathic pain [42, 62, 68, 69].
留言 (0)