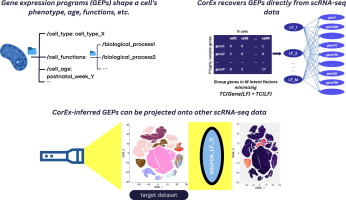
Upon exposure to internal or external signals, genes are precisely coordinated to induce different cellular transcriptional states, thus providing invaluable insights into the co-regulation of genes in diverse cell types, tissues, and organisms. These co-regulated gene sets give rise to distinct biological phenotypes and functions, commonly referred to as gene expression programs (GEPs). Next Generation Sequencing (NGS) methods such as RNA sequencing allow us to measure these programs to unveil the biological processes behind them. Traditionally, gene expression studies were performed on bulk populations of cells, which potentially obscure heterogeneity within the population. The more recent development of single-cell RNA sequencing (scRNA-seq) technology has revolutionized our understanding of gene expression by allowing for the analysis of GEPs in individual cells. This provides a much more detailed and specific understanding of cellular heterogeneity and gene expression dynamics.
While scRNA-seq technology has transformed our understanding of gene expression and cellular heterogeneity, there are still significant challenges associated with analyzing this data. These challenges include accurate cell type identification, technical variation, detection of genes with low expression, and the potential for bias or noise in the data analysis [1]. Advances in methods for dimensionality reduction, clustering, identification of differentiation paths, and cell–cell communication have helped address some of those challenges [2], [3], [4], [5]. These methods predominantly focus on clustering cells and identifying their phenotypes but do not address other critical features of a cell’s gene expression state, such as response to external stimuli or treatment [6], [7]. This represents a significant issue, as one cell can participate in multiple biological processes characterized by different GEPs. For example, a cell can have the phenotype of a CD8 + T cell, undergo antigen-mediated activation, and receive an external signal that induces exhaustion. Here, the gene set that characterizes the CD8 + T cell type is called the identity GEP, and the gene sets associated with antigen recognition and exhaustion are activity GEPs. Identity GEPs are cell specific whereas activity GEPs may be expressed by multiple cell types performing similar functions. The ability to identify functional programs is critical to improving the understanding of disease processes on the single-cell level.
The identification of activity GEPs in scRNA-seq data, however, has proved difficult. Current methods most commonly utilize gene set enrichment analysis (GSEA) applied to differentially expressed genes in contrasting samples [8], [9]. These methods suffer from the limitations of batch effect, limited contrasting tissue samples, and high cost when applied to scRNA-seq studies. However, if a group of cells in scRNA-seq data express a specific activity GEP, there is the potential to directly extract GEPs from the data without a control sample. Decomposition methods, such as Principal Component Analysis (PCA), Independent Component Analysis (ICA), and Non-Negative Matrix Factorization (NMF), which are typically used for dimensionality reduction, can be applied to obtain GEPs from scRNA-seq data [10], [11], [12], [13]. PCA and ICA, however, do not perform well when applied to finding biologically meaningful activity GEPs due to inherent aspects of the methods. PCA can be used to distinguish different cell types among a large group of heterogeneous cells, however to accomplish this the method maximizes the variability of specific factors, which may lead to mixing the signal from multiple biological processes in a single component [14]. ICA is powerful when used to separate independent sources that are mixed together in observed data, but requires the assumption that the sources are non-Gaussian and linearly mixed, which may not hold in practice, limiting its applicability to biologic data.
Out of these dimensionality reduction methods, NMF has shown some success in retrieving activity GEPs. Puram, et al. applied NMF to scRNA-seq data of head and neck squamous carcinoma malignant cells and discovered GEPs related to cell cycle, stress, hypoxia, epithelial differentiation, and partial epithelial-to-mesenchymal transition [15]. Some variants of NMF, such as scCoGAPS and consensus NMF (cNMF), were specifically developed for discovery of GEPs in scRNA-seq data [16], [17]. cNMF is a method of GEP identification based on a meta-analysis of NMF decomposition results. The developers of cNMF showed that their method can find activity GEPs associated with hypoxia, the cell cycle, and reaction to depolarization. scCoGAPS uses NMF to decompose a scRNA-seq matrix into Pattern and Amplitude matrices. The method employs an atomic prior, a specific type of prior distribution, to capture three biological constraints: non-negativity, sparsity, and smoothness. In their work, Stein-O’Brien et al. demonstrated that scCoGAPS was capable of finding identity and activity GEPs in developing mouse retina scRNA-seq data [16]. These NMF-based approaches, however, have a number of limitations. First, as the input matrix must be positive, it cannot represent repressed genes. Second, each cell is represented as a linear combination of GEPs when using NMF. This approach, therefore, may not be able to effectively interpret the biologically plausible scenario where the expression of a specific GEP results in decreased expression of a different GEP in the same cell.
In this study, we demonstrate that biologically meaningful GEPs can be inferred directly from scRNA-seq data by grouping genes based on total correlation optimization function using the unsupervised, machine learning approach linear CorEx. First, using simulated data, we show that linear CorEx identifies cell types and activity programs in scRNA-seq data and demonstrates better predictive power when compared to other methods. Second, using real scRNA-seq data from the mouse dentate gyrus, we find biologically meaningful GEPs associated with different cell types, developmental ages, and cell cycle programs. We also demonstrate the potential for improved applicability and throughput of this method using transfer learning of inferred GEPs across samples using another scRNA-seq dataset from the adult mouse dentate gyrus. Additionally, linear CorEx showed cross-species sensitivity when using transfer learning to project mouse dentate gyrus-derived GEPs to human dentate gyrus data. Furthermore, to demonstrate CorEx's capacity to uncover biologically putative GEPs across diverse tissues and conditions, we apply it to mouse embryonic colon development data. Subsequently, we project the resulting GEPs onto a colitis dataset to discern disease-specific programs. Together the techniques in this study show that linear CorEx has the ability to overcome some significant issues related to current scRNA-seq analyses and represents a step forward for the understanding of biological processes at the single-cell level.




留言 (0)