Obstructive sleep apnoea (OSA) arises from repetitive closure of the upper airway during sleep, resulting in daytime somnolence and heightened cardiovascular risk. A major contributor to this risk (and increased mortality) is the onset of hypertension, which arises with high prevalence in OSA. The strong association between OSA and hypertension has instigated a substantial body of research. Long-term follow-up in patients with disordered sleep finds a significant proportion develop hypertension in dose–response manner, suggesting a causal relationship [1]. Apnoea results in a chemoreflex-induced activation of sympathetic nerves, triggering an acute haemodynamic response [2]. The response starts with widespread vasoconstriction and a rise in systemic and pulmonary vascular resistance and pressures. When breathing resumes, a transient increase in venous return and cardiac output further increases arterial pressures. Finally, electroencephalographic microarousals further contribute to sympathetic stimulation and blood pressure. Repeated cycles of the above are believed to induce chronic hypertension in OSA, via the activation of several maladaptive processes including inflammation, dysfunction of the endothelium, oxidative stress, vasoactive factors (e.g. endothelin), and metabolic changes [3]. The reduced sleep duration arising from OSA has also been linked to hypertension [4].
Accordingly, there was a clear mechanistic rationale for trialling CPAP as a novel antihypertensive treatment. Although early studies suggested considerable promise of continuous positive airway pressure (CPAP) as an antihypertensive (reductions as high as ∼10 mmHg), the subsequent major body of evidence, however, indicate a more modest treatment effect (2–3 mmHg), and which could imply that many patients could have other primary drivers of hypertension (e.g. essential hypertension) separate to OSA. As a result, it has been proposed that greater antihypertensive effects could be achieved by selecting patients with features marking them as ‘responders’. Predicting blood pressure response to CPAP would be advantageous, in allowing clinicians to select patients who derive most benefit. Greater BP response to CPAP has been reported in younger patients, and in patients with poor hypertensive control, higher daytime sleepiness, and greater severity of sleep apnoea.
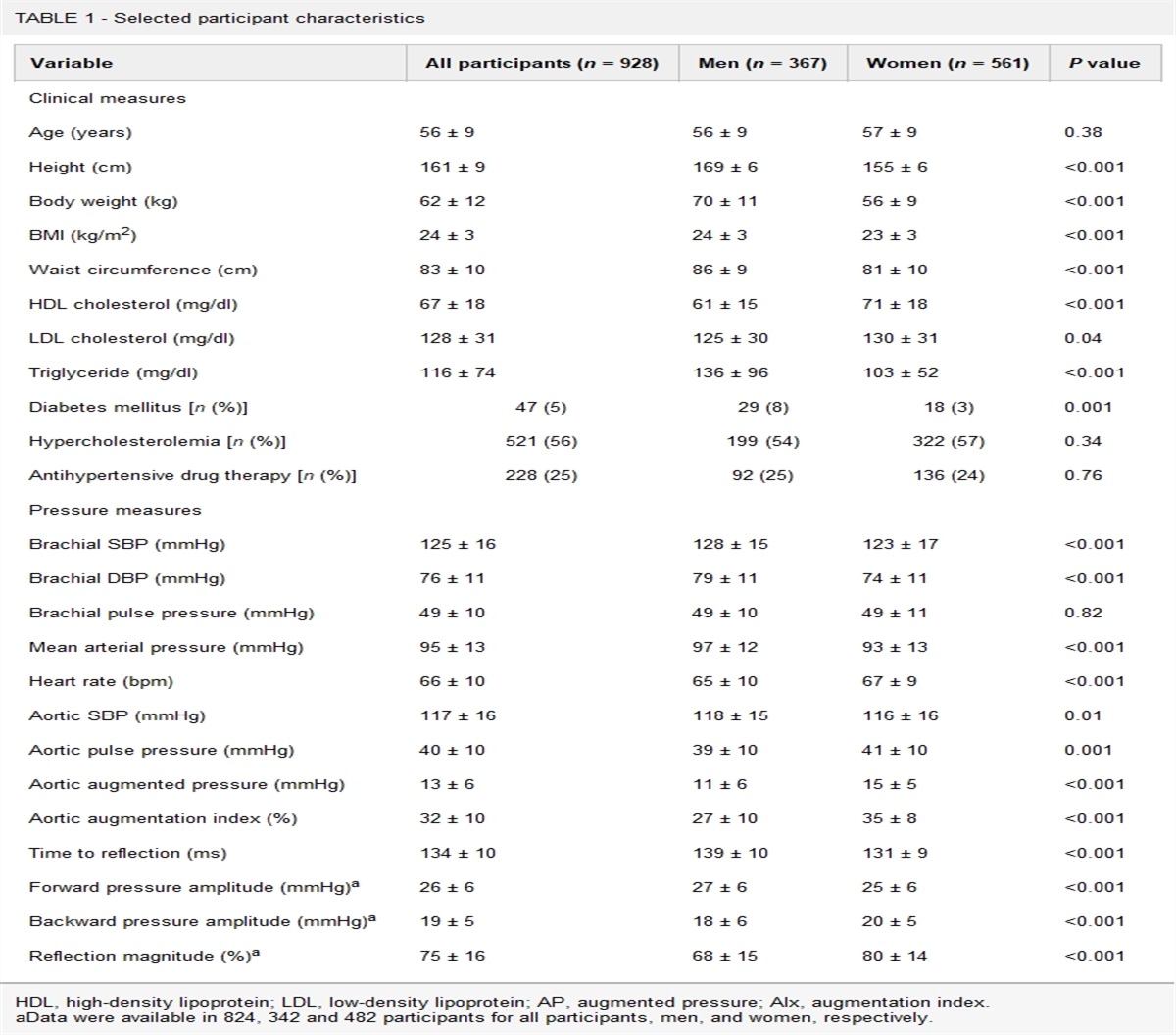
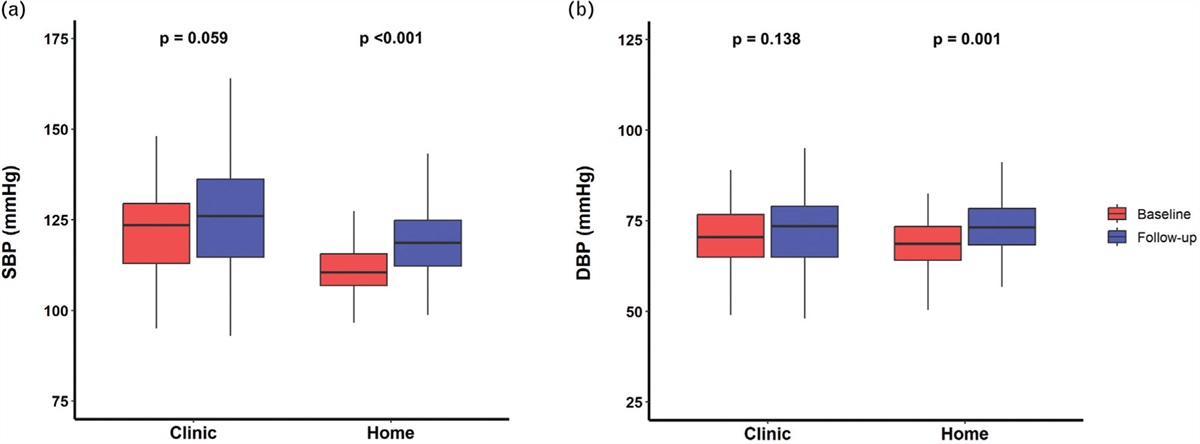
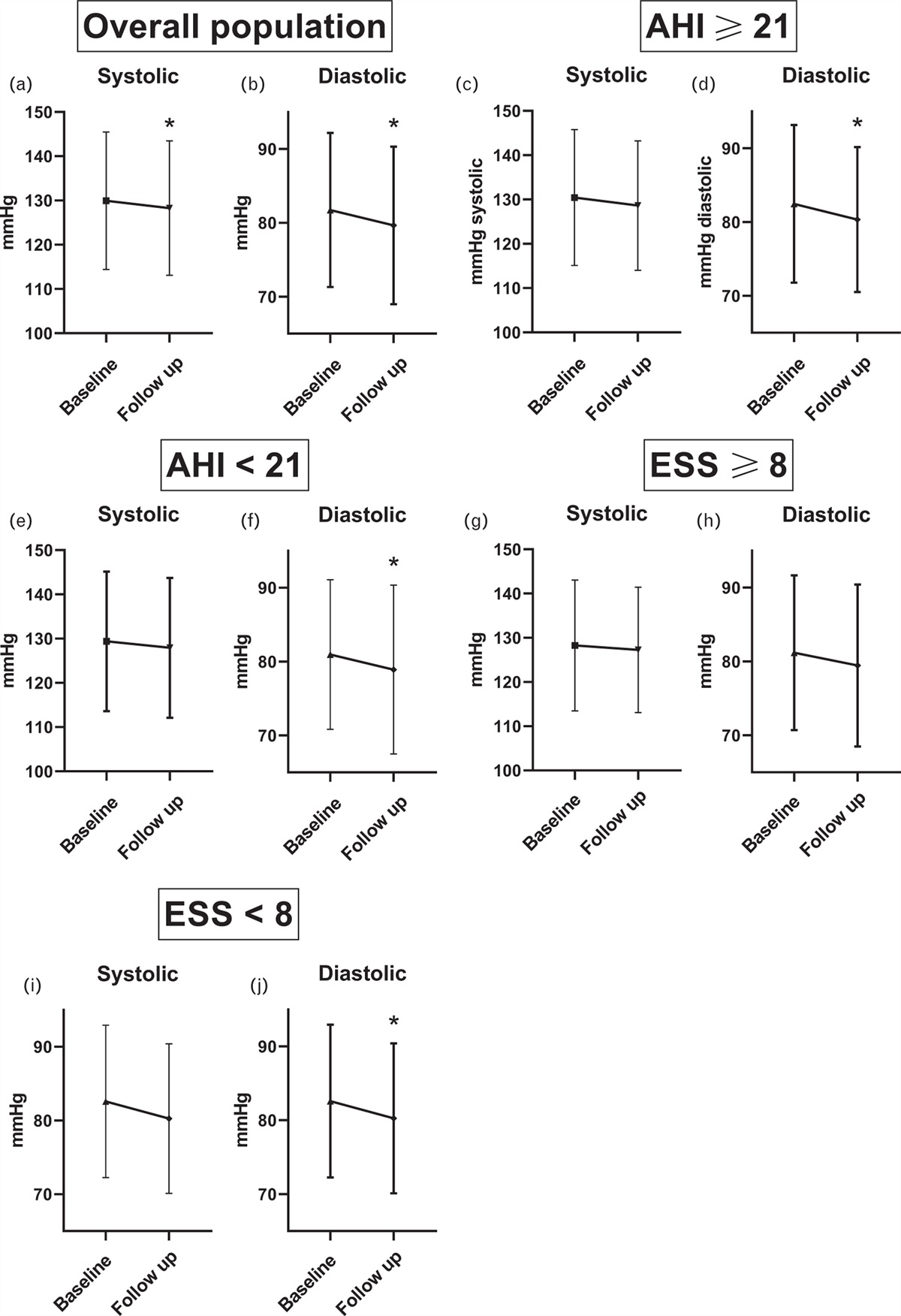
In an attempt to identify blood pressure responders, Seidel et al. have designed and implemented a robust study, using the baseline apnoea hypopnea index (AHI) and the extent of daytime sleepiness (ESS), as tools to assess the severity of OSA. They sought to address whether these scores could predict which patients develop greatest antihypertensive responses to CPAP. The key findings from this comprehensive study of 555 patients with obstructive sleep apnoea are two-fold. First, in keeping with the wider literature, nocturnal CPAP resulted in a very mild BP-lowering effect. Second, neither AHI or ESS were effective in predicting more substantial BP decreases (≥5 mmHg) [5]. The authors are commended for significant patient numbers, completeness of data sets, and long duration of follow-up (∼1 year). In combination, these allow for a robust assessment of long-term treatment effects of CPAP, and provides valuable evidence of modest daytime BP reductions across the range of AHI and ESS scores. Nevertheless, several points must be considered.
Firstly, it is interesting that there was minimal difference in BP response across the range of AHI and ESS scores, and this is unexplained. The cohort was notable in having well controlled baseline BP (whereas greatest benefit from CPAP has been reported in patients with poorly controlled hypertension [3]), and it is possible that BP was too low to elicit large treatment responses. The fall in BP may have been so small that the study had insufficient power to detect subtle differences between groups. Alternatively, the chronicity of hypertension-OSA could also impact on the treatment response to OSA. Here the majority of the study population (72%) had an established diagnosis of hypertension (and were already taking antihypertensives). In future work, it would be interesting to establish whether CPAP has a preventive antihypertensive effect when started before onset of OSA hypertension (and before the induction of chronic maladaptive processes). It would also be valuable to examine whether AHI and ESS can identify those patients who can derive most benefit from CPAP under these circumstances. It should also be noted that some patients may have underlying essential hypertension, independent of OSA, which may dampen average effects of CPAP on blood pressure.
Secondly, as the authors allude, the long follow-up period does introduce potential for other biases. Weight, lifestyle habits, antihypertensive treatments and intercurrent illnesses may have all changed over the period, which can modify both BP and scores of sleepiness and apnoea. Nevertheless only a minority of patients had a change in antihypertensive treatment over the period, and even when these were excluded, the same conclusions remained.
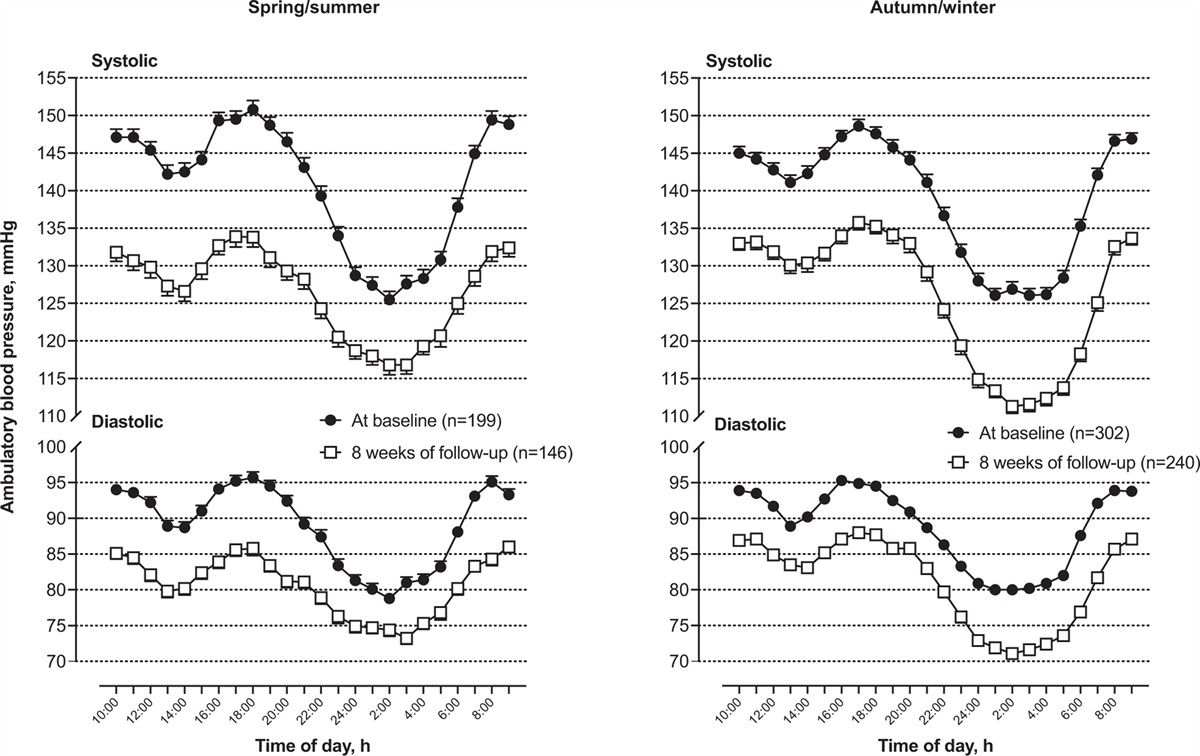
Thirdly, the retrospective study relied on single office BP measurements and did not have data on nocturnal BP. Although OSA is a known cause of daytime hypertension, one would nevertheless expect nocturnal treatment effects to be greater, including effects on nocturnal nondipping blood pressure.
To conclude, the work by Seidel et al. provides a valuable addition to the body of evidence predicting CPAP blood pressure responses, and suggests that in patients with established OSA, neither severity of apnoea nor daytime sleepiness can predict effect on BP. As things stand, CPAP appears to be modest as a standalone treatment of OSA-hypertension across the range of AHI and ESS scores. These findings should be corroborated in OSA across a range of follow-up intervals and patient subgroups, including those patients with poor hypertensive control, and in long-term follow-up of OSA patients before onset of hypertension. Studies should also include nocturnal BP assessments.
ACKNOWLEDGEMENTS
Conflicts of interest
AMH receives lecture fees from Servier. DCH has no conflicts of interest.
REFERENCES
1. Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000; 342:1378–1384.
2. Shamsuzzaman AS, Gersh BJ, Somers VK. Obstructive sleep apnea: implications for cardiac and vascular disease. JAMA 2003; 290:1906–1914.
3. Kapa S, Sert Kuniyoshi FH, Somers VK. Sleep apnea and hypertension: interactions and implications for management. Hypertension 2008; 51:605–608.
4. Gangwisch JE, Heymsfield SB, Boden-Albala B, Buijs RM, Kreier F, Pickering TG, et al. Short sleep duration as a risk factor for hypertension: analyses of the first National Health and Nutrition Examination Survey. Hypertension 2006; 47:833–839.
5. Pengo MF, Soranna D, Giontella A, Perger E, Mattaliano P, Mattaliano PG, et al. Obstructive sleep apnoea treatment and blood pressure: which phenotypes predict a response? A systematic review and meta-analysis. Eur Respir J 2020; 55.


留言 (0)