Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium, BUPGEN, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, 23andMe Research Team, Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E et al (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51:431–444
Barthélémy, Caron Yufei, Luo Antonio, Rausell (2019) NCBoost classifies pathogenic non-coding variants in Mendelian diseases through supervised learning on purifying selection signals in humans Genome Biol 20(1). https://doi.org/10.1186/s13059-019-1634-2
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M et al (2010) The NIH roadmap epigenomics mapping consortium. Nat Biotechnol 28:1045–1048
Article
CAS
PubMed
PubMed Central
Google Scholar
Buitinck L, Louppe G, Blondel M, Pedregosa F, Mueller A, Grisel O, Niculae V, Prettenhofer P, Gramfort A, Grobler J, Layton R (2013) API design for machine learning software: experiences from the scikit-learn project. arXiv preprint arXiv:1309.0238
Carter H, Douville C, Stenson PD, Cooper DN, Karchin R (2013) Identifying mendelian disease genes with the variant effect scoring tool. BMC Genomics 14:S3
Article
PubMed
PubMed Central
Google Scholar
Chen K, Zhao H, Yang Y (2022a) Capturing large genomic contexts for accurately predicting enhancer-promoter interactions. Brief Bioinform 23:bbab577
Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, Alföldi J, Watts NA, Vittal C, Gauthier LD, Poterba T, Wilson MW et al (2023) A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625:92–100
Cheng J, Nguyen TYD, Cygan KJ, Çelik MH, Fairbrother WG, Avsec Ž, Gagneur J (2019) MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol 20:48
Article
PubMed
PubMed Central
Google Scholar
Elkon R, Agami R (2017) Characterization of noncoding regulatory DNA in the human genome. Nat Biotechnol 35:732–746
Article
CAS
PubMed
Google Scholar
Fabian P (2011) Scikit-learn: Machine learning in Python. J Mach Learn Res 12:2825
Felsenstein J, Churchill GA (1996) A Hidden Markov Model approach to variation among sites in rate of evolution. Mol Biol Evol 13:93–104
Article
CAS
PubMed
Google Scholar
Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, Gal Y, Marks DS (2021) Disease variant prediction with deep generative models of evolutionary data. Nature 599:91–95
Article
CAS
PubMed
Google Scholar
Genome Interpretation Consortium (2022) CAGI, the critical assessment of genome interpretation, establishes progress and prospects for computational genetic variant interpretation methods. ArXiv E-Prints arXiv-2205.
Gerasimavicius L, Livesey BJ, Marsh JA (2022) Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat Commun 13:3895
Article
CAS
PubMed
PubMed Central
Google Scholar
Guyon I, Weston J, Barnhill S, Vapnik V (2002) Gene selection for cancer classification using support vector machines. Mach Learn 46:389–422
Hanson J, Yang Y, Paliwal K, Zhou Y (2017) Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Abs Bioinform 33(5):685–692. https://doi.org/10.1093/bioinformatics/btw678
He K, Zhang X, Ren S, Sun J (2016) Deep residual learning for image recognition. In: Proceedings of the IEEE conference on computer vision and pattern recognition
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC et al (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99:877–885
Article
CAS
PubMed
PubMed Central
Google Scholar
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G (2016) M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 48:1581–1586
Article
CAS
PubMed
Google Scholar
Ke Y, Rao J, Zhao H, Lu Y, Xiao N, Yang Y (2020) Accurate prediction of genome-wide RNA secondary structure profile based on extreme gradient boosting. Bioinformatics 36:4576–4582
Article
CAS
PubMed
Google Scholar
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at UCSC. Genome Res 12(6):996–1006
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315
Article
CAS
PubMed
PubMed Central
Google Scholar
Kircher M, Xiong C, Martin B, Schubach M, Inoue F, Bell RJA, Costello JF, Shendure J, Ahituv N (2019) Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat Commun 10:3583
Article
PubMed
PubMed Central
Google Scholar
Laskowski RA, Stephenson JD, Sillitoe I, Orengo CA, Thornton JM (2020) VarSite: disease variants and protein structure. Protein Sci 29:111–119
Article
CAS
PubMed
Google Scholar
Li C, Zhi D, Wang K, Liu X (2022) MetaRNN: differentiating rare pathogenic and rare benign missense SNVs and InDels using deep learning. Genome Med 14:115
Article
PubMed
PubMed Central
Google Scholar
Livingstone M, Folkman L, Yang Y, Zhang P, Mort M, Cooper DN, Liu Y, Stantic B, Zhou Y (2017) Investigating DNA-, RNA-, and protein-based features as a means to discriminate pathogenic synonymous variants: LIVINGSTONE et al. Hum Mutat 38:1336–1347
Mendez MF (2019) Early-onset Alzheimer disease and its variants. Contin Lifelong Learn Neurol 25:34–51
Article
Google Scholar
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20:110–121
Article
CAS
PubMed
PubMed Central
Google Scholar
Qi H, Zhang H, Zhao Y, Chen C, Long JJ, Chung WK, Guan Y, Shen Y (2021) MVP predicts the pathogenicity of missense variants by deep learning. Nat Commun 12:510
Article
CAS
PubMed
PubMed Central
Google Scholar
Raimondi D, Tanyalcin I, Ferté J, Gazzo A, Orlando G, Lenaerts T, Rooman M, Vranken W (2017) DEOGEN2: prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res 45:W201–W206
Article
CAS
PubMed
PubMed Central
Google Scholar
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47:D886–D894
Article
CAS
PubMed
Google Scholar
Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518:317–330
Article
PubMed Central
Google Scholar
Shwartz-Ziv R, Armon A (2022) Tabular data: deep learning is not all you need. Inf Fusion 81:84–90
Article
Google Scholar
Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, Rowe LD, Dreszer TR et al (2016) ENCODE data at the ENCODE portal. Nucleic Acids Res 44:D726–D732
Article
CAS
PubMed
Google Scholar
Smedley D, Schubach M, Jacobsen JOB, Köhler S, Zemojtel T, Spielmann M, Jäger M, Hochheiser H, Washington NL, McMurry JA, Haendel MA, Mungall CJ et al (2016) A whole-genome analysis framework for effective identification of pathogenic regulatory variants in Mendelian disease. Am J Hum Genet 99:595–606
Article
CAS
PubMed
PubMed Central
Google Scholar
Sundaram L, Gao H, Padigepati SR, McRae JF, Li Y, Kosmicki JA, Fritzilas N, Hakenberg J, Dutta A, Shon J, Xu J, Batzoglou S et al (2018) Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 50:1161–1170
Article
CAS
PubMed
PubMed Central
Google Scholar
Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B (2014) Synonymous mutations frequently act as driver mutations in human cancers. Cell 156:1324–1335
Article
CAS
PubMed
Google Scholar
Valette K, Li Z, Bon-Baret V, Chignon A, Bérubé J-C, Eslami A, Lamothe J, Gaudreault N, Joubert P, Obeidat M, van den Berge M, Timens W et al (2021) Prioritization of candidate causal genes for asthma in susceptibility loci derived from UK Biobank. Commun Biol 4:700
Article
CAS
PubMed
PubMed Central
Google Scholar
Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, Chang HY (2014) Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505:706–709
Article
CAS
PubMed
PubMed Central
Google Scholar
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Article
PubMed
PubMed Central
Google Scholar
Wang T, Ruan S, Zhao X, Shi X, Teng H, Zhong J, You M, Xia K, Sun Z, Mao F (2021) OncoVar: an integrated database and analysis platform for oncogenic driver variants in cancers. Nucleic Acids Res 49:D1289–D1301
Article
CAS
PubMed
Google Scholar
Wu Y, Liu H, Li R, Sun S, Weile J, Roth FP (2021) Improved pathogenicity prediction for rare human missense variants. Am J Hum Genet 108:1891–1906
Article
CAS
PubMed
PubMed Central
Google Scholar
Zappala Z, Montgomery SB (2016) Non-coding loss-of-function variation in human genomes. Hum Hered 81:78–87
Article
CAS
PubMed
Google Scholar
Zhao H, Yang Y, Lin H, Zhang X, Mort M, Cooper DN, Liu Y, Zhou Y (2013) DDIG-in: discriminating between disease-associated and neutral non-frameshifting micro-indels. Genome Biol 14:R23
Article
PubMed
PubMed Central
Google Scholar

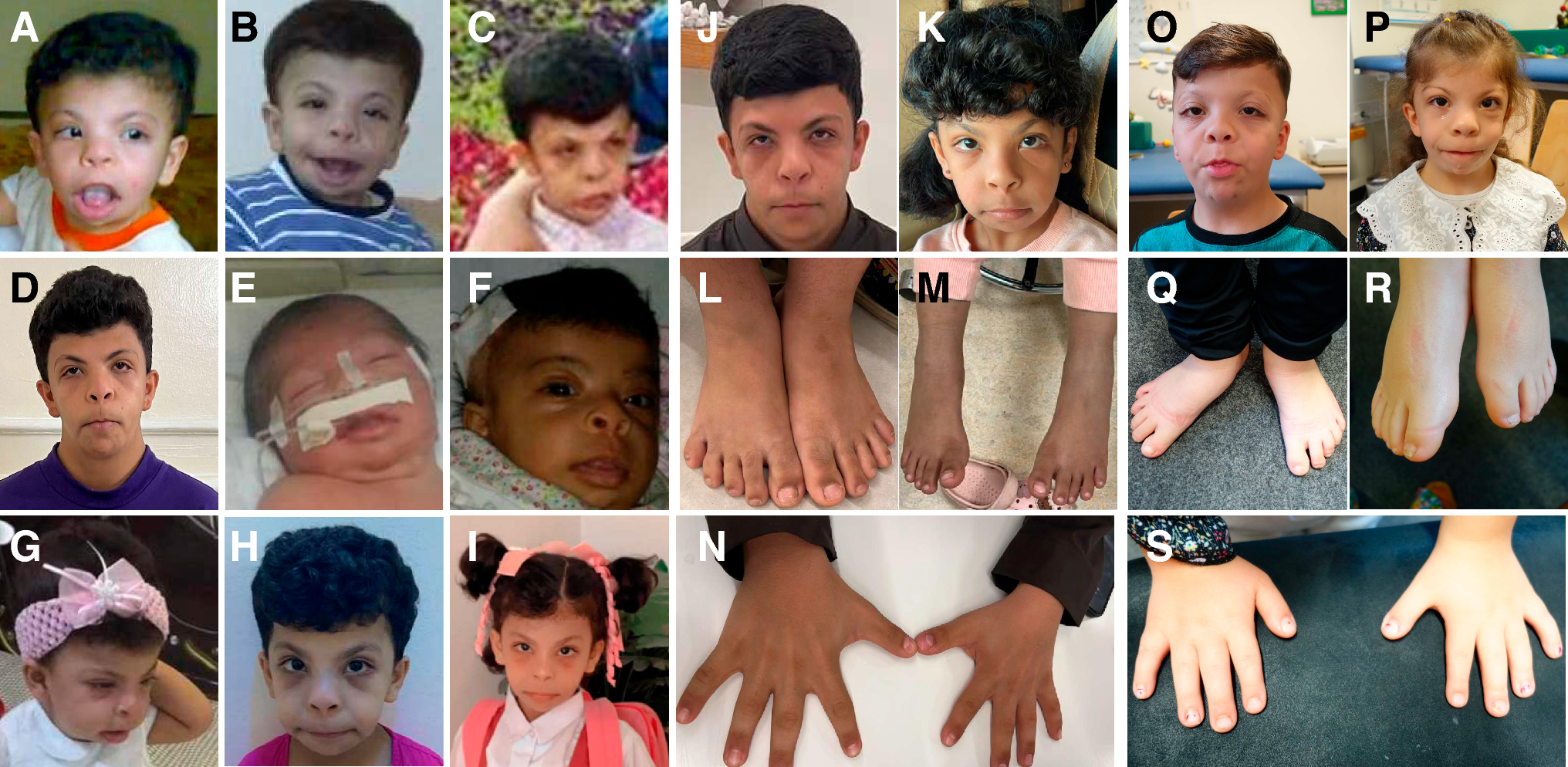
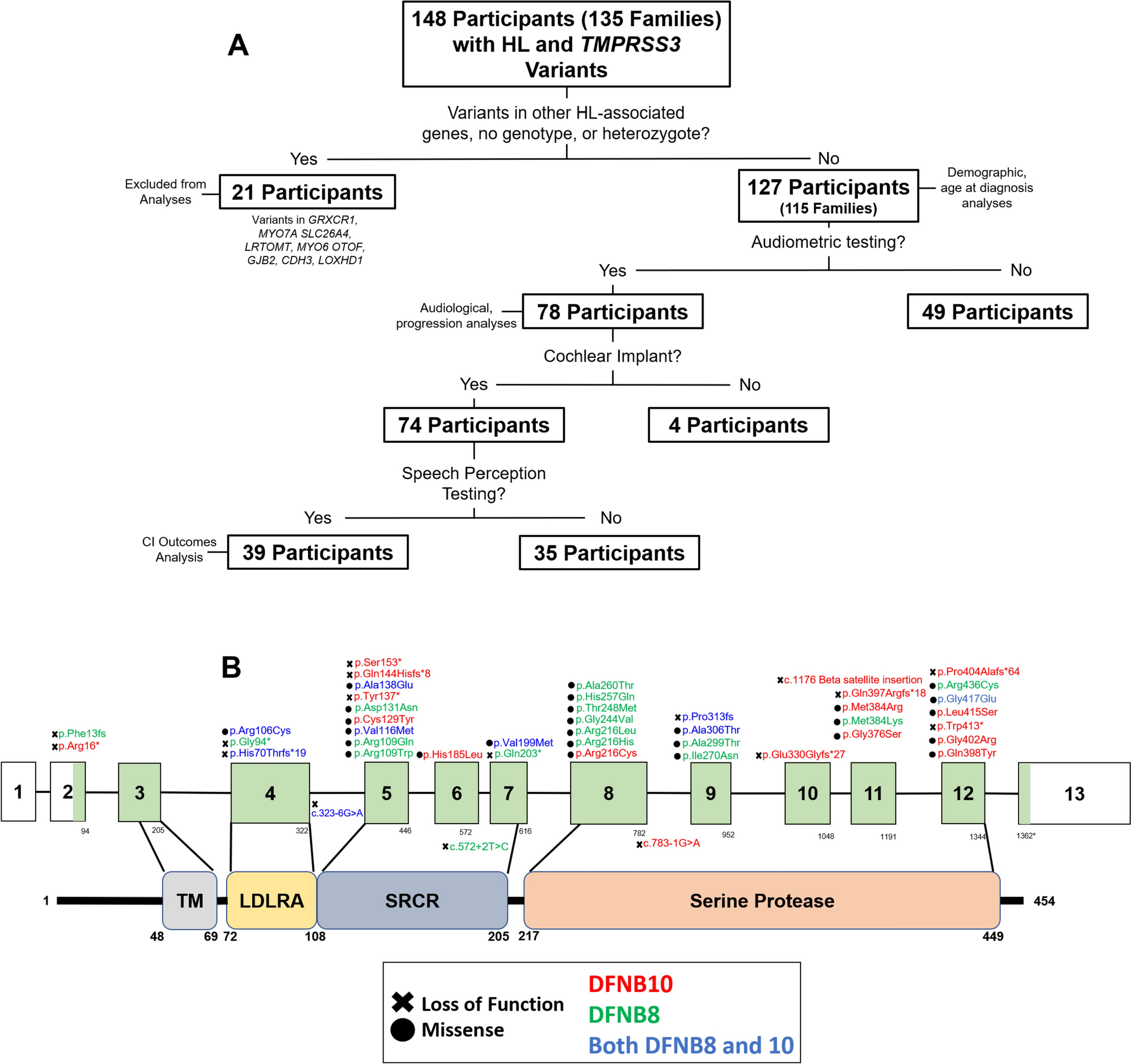
留言 (0)