Type-I NF1 microdeletion mainly results from a germline NAHR, unlike the other classes of NF1 microdeletion that arise from recombination events that also occur at post-zygotic level (reviewed by Kehrer-Sawatzki et al. 2017). Mosaicism can be involved in expression variability of the clinical phenotype in a fraction of NF1 microdeletion patients, particularly those with type-II and atypical NF1 deletions. In contrast, type-I NF1 microdeletion are predominantly germline deletions. It is still unclear whether the clinical phenotype associated with type-I NF1 microdeletion is mainly caused by the haploinsufficiency of the genes located within the type-I NF1 microdeletion interval or whether additional pathogenetic mechanisms contribute to the clinical expression. To investigate the NF1 microdeletion syndrome etiopathogenesis, we enrolled 22 type-I NF1 microdeletion syndrome patients. The incidence of the clinical signs, included in the classical NF1 phenotype or the typical ones of the microdeletion syndrome, in our cohort is comparable to that reported in the literature (reviewed by Kehrer-Sawatzki et al. 2017), allowing us to consider our case studies as sufficiently representative of the clinical picture of type-I NF1 microdeletion syndrome.
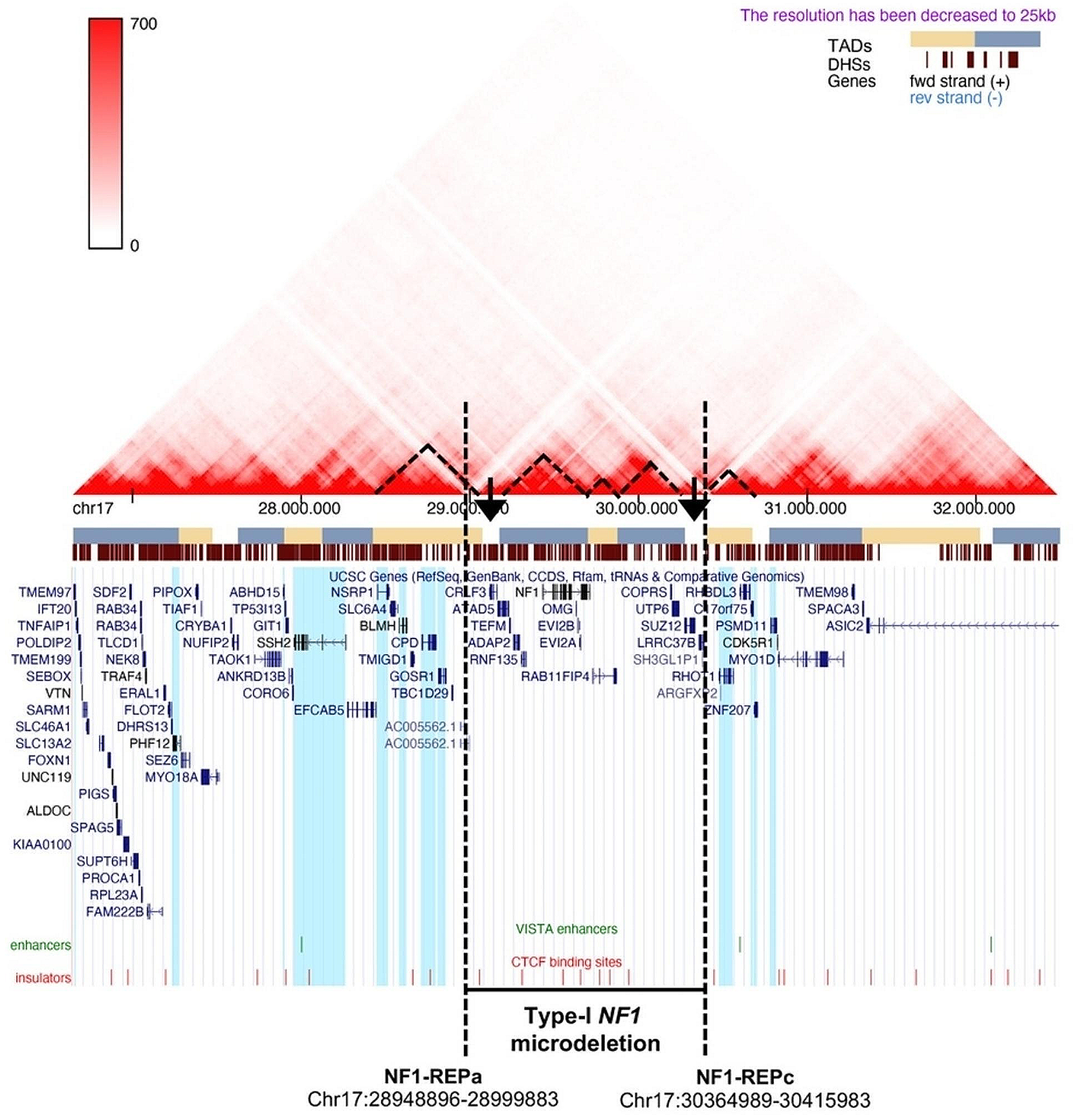
Position effect on expression of genes flanking deletions has been poor studied in microdeletion syndromes and has only been described in one NF1 microdeletion syndrome patient (Ferrari et al. 2017; Tritto et al. 2023b). We performed in silico analysis of 17q11.2 TADs on a single lymphoblastoid cell line, in accordance with the subsequent real time analysis on retrotranscribed cDNA derived from patient whole blood RNA. Nevertheless, 3D Genome Browser shows that the chromatin topological organization of 17q11.2 region is fairly conserved in other cell lines, such as NHEK (normal human epidermal keratinocytes), DLPFC (dorsolateral prefrontal cortex) and GZ (germinal zone of human cerebral cortex) (Supplementary Fig. S4). Type-I NF1 microdeletion syndrome patients, losing the same TADs, should be subjected to the expression dysregulation of the same genes that should be involved in the onset of common abnormal clinical traits.
We demonstrated the position effect in peripheral blood analyzing genes ubiquitously expressed, therefore their expression is possibly affected in other tissues, even if the regulatory elements may vary in cells derived from other tissues. We found an upregulation of GOSR1, CPD, NSRP1, and PHF12 genes and a downregulation of RHOT1, c17orf75, and ZNF207 genes, never correlated to NF1 microdeletion syndrome, at our knowledge. Of note, three out of the four over-expressed genes belong to the partially deleted TAD, upstream of the deletion, and the three hypo-expressed genes map to the first telomeric TAD following the deletion, leading us to speculate that underlying the position effect may be a mechanism of enhancer competition (Kleinjan and van Heyningen 1998) which is restricted to the new putative TAD, presumably generated as a result of the deletion. Genetic alterations that change the architecture of TADs, resulting in non-canonical enhancer-gene interactions that increase the expression of the interacting genes and lead to decreased expression of genes they normally regulate, causing severe phenotypes, have been reported previously. Lupiáñez and colleagues described how heterozygous deletions on chromosome 2q35-36 that alter the higher-order chromatin organization of the WNT6/IHH/EPHA4/PAX3 locus can allow interactions between a cluster of limb enhancers, normally associated with EPHA4, and the PAX3 promoter region, leading to an ectopic limb expression of PAX3 and causing brachydactyly (Lupiáñez et al. 2015).
The limitation of in silico analysis of TADs is that they may vary based on the different methods of identification (Dang et al. 2023), but our results are supported by the deregulation of flanking genes expression, which is restricted to the two partially deleted TADs, with the exception of one gene.
Given that gene expression regulation is a complex biological process, resulting from the activity of several mechanisms, a different modulation of gene activity due to the position effect might impact the penetrance or severity degree of a specific clinical trait shared by type-I NF1 microdeletion patients. Notably, there may be correlations between the expression deregulation of the RHOT1 and ZNF207 genes and some phenotypic features observed in type-I NF1 microdeletion syndrome patients. RHOT1 maps 70 Kb far from the telomeric breakpoint of type-I NF1 microdeletion. RHOT1 encodes MIRO1, a protein belonging to the Rho GTPases and reported to be involved in mitochondrial transport (Fransson et al. 2003) and in migration and polarity of lymphocytes (Morlino et al. 2014). MIRO1 is known to interfere with mitochondrial quality control and Mitochondria-ER contact sites (MERCS). Low expression levels of RhoT1 appear to be significantly associated with lymph node metastasis and shorter survival in pancreatic cancer patients. Interestingly, pancreatic endocrine tumors occur in patients with NF1 and have been reported to be a cause of death in these patients (Khosrotehrani et al. 2003; Jensen et al. 2008). Deregulation of RHOT1 expression may be a risk factor for pancreatic cancer in NF1 microdeletion patients, although the reported data refer to NF1 patients without distinguishing between different forms of NF1. MERCs are known to be involved in the regulation of many cellular functions (calcium homeostasis, lipid metabolism, autophagy, and apoptosis) with a broad implication into synaptic events, due to the presence of these contacts in various parts of neurons and glial cells. Despite MERCs function is not fully explored, it is intriguing to observe a downregulation of a gene strictly related to MERCs in this peculiar class of patients, with a typical neurodevelopmental involvement (Shirokova et al. 2020). ZNF207 is located 300 Kb distant from the telomeric breakpoint of type-I NF1 microdeletion. This gene encodes for BuGZ, a zinc finger protein that binds to spindle microtubules and regulates chromosome alignment during mitosis (Jiang et al. 2014). SiRNA-mediated knockdown of ZNF207, carried out on human embryonic stem cells (hESCs), has shown that reduction of its gene expression impairs self-renewal and pluripo(Finn et al. 2019)tency of these cells by blocking ectoderm differentiation (Fang et al. 2018). Because nervous tissue and epidermis, both widely affected by the symptoms of NF1 microdeletion syndrome, derive from the ectoderm, hypo-expression of ZNF207 could be a candidate mechanism for playing a role in the altered development of these tissues, although further investigation is needed to confirm this hypothesis.
To identify the putative chromatin remodeling causative of the position effect found in microdeletion patients, a 4C-Seq assay was carried out on samples from three type-I NF1 microdeletion patients and three controls. The analysis was performed on the two pools of patients and healthy donors to minimize the biological variability, possibly due to cellular and allelic differences (Finn et al. 2019). Interactions between the selected viewpoint, represented by the RHOT1 promoter located after the telomeric deletion breakpoint, and the whole genome, which differed in patients and controls, confirmed an altered chromatin folding of the chromosomal region 17q11.2 in our patients.
Interestingly, in the NF1 microdeletion patients, the gain of a new interaction between the viewpoint and the SLC6A4 gene, mapped to the centromeric TAD partially deleted and found to be hyper-expressed by qPCR in the peripheral blood of the 15 patients, was identified. This evidence further suggests that the deletion may lead to the formation of a new TAD, given by the fusion between the centromeric and telomeric TADs, which may be responsible for the change in the regulatory landscape acting in cis on genes showing aberrant expression.
The loss of interactions between the viewpoint and the sequences contained in the chromosome region within the deletion shown by the 4C-seq data revealed that chromatin remodeling occurs not only in the region flanking the deletion, but also in the deletion interval, on the non-deleted homologous chromosome. This evidence suggests that the presence of the deletion may lead to a change in the chromatin structural organization of the homologous chromosome, indicating the involvement of trans-acting mechanisms. This hypothesis is supported by the higher expression of some genes within the deletion interval in our microdeletion patients compared to the expected 50% reduction.
Furthermore, the loss of DNA-DNA interactions within the type-I NF1 deletion leads us to speculate on a possible crosstalk between the two chromosomes. Although to our knowledge the occurrence of this molecular mechanism as a result of a microdeletion has not been previously described, some evidence supports the hypothesis of crosstalk between homologous chromosomes, even during the interphase of the cell cycle. The transcription machinery tends to co-express the two alleles of the same gene, located on the two homologous chromosomes, almost simultaneously (Santoni et al. 2017). In addition, chromosomes preferentially occupy defined regions in the cell nucleus, which are known as chromosomal territories, which represent an additional level of gene expression regulation (Bolzer et al. 2005). Changes in the three-dimensional conformation of specific chromosomes and the area of their chromosomal territories, due to chromosomal rearrangements, have been observed in the interphase DNA of spermatozoa, by two-dimensional fluorescent in situ hybridization (FISH) performed both in spermatozoa with normal chromosome arrangement and in those characterized by chromosomal imbalance (Mebrek et al. 2020). The results obtained from Mebrek’s study demonstrate that nuclear architecture has a fragile organization and that chromosomal abnormalities can affect the entire nucleus. Understanding the molecular basis leading to the chromatin remodeling, observed on the non-deleted chromosome in patients with type-I NF1 microdeletion, may allow the identification of a pathogenetic mechanism common to other microdeletion syndromes.
Most of the genes included in the type-I NF1 microdeletion interval have been identified as dosage-sensitive genes in previous studies (reviewed by Kehrer-Sawatzki et al. 2017). By checking the constraint metric probability of being LoF intolerant (pLI), according to gnomAD v.2.1.1 database, the 14 deleted genes could be classified into three categories: (1) LoF intolerant, characterized by a pLI ≥ 0.9 (5/14, 36%), including the ATAD5, NF1, OMG, RAB11FIP4, and SUZ12 genes, whose haploinsufficiency could lead to pathological consequences in patients with type-I NF1 microdeletion, thus worsening their phenotype compared to that of NF1 patients without the microdeletion; (2) likely haplosufficient genes with 0.1 < pLI < 0.9 (2/14, 14%), including COPRS and TEFM; (3) LoF tolerant genes with a pLI ≤ 0.1 (7/14, 50%), namely, ADAP2, CRLF3, EVI2A, EVI2B, LRRC37B, RNF135, and UTP6, for which the pLI metric suggests the existence of a different pathomechanism, e.g., a two-hit recessive model, in contributing to the patients’ complex phenotype (Supplementary Fig. S5 and Table S2). Consistently, deletion of ATAD5, NF1, RAB11FIP4, SUZ12, and COPRS genes n to may increase tumor risk in NF1 microdeletion patients (as reported by Kehrer-Sawatzki et al. 2017), while OMG deletion gene has been previously associated to cognitive impairment in this NF1 patients’ subgroup (Venturin 2004). Heterozygous deletion of the TEFM gene, whose product acts as a general elongation factor for mtDNA transcription in mammalian mitochondria (Minczuk et al. 2011), does not impair mitochondrial function of mtDNA replication and transcription (Minczuk et al. 2011), suggesting that its hemizygosity is unlike affecting the phenotype of NF1 microdeletion patients. Of note, among the genes predicted to be LoF tolerant, ADAP2 and RNF135 were found to be associated to cardiovascular malformations, overgrowth, and dysmorphisms by functional studies (Douglas et al. 2007; Venturin et al. 2014). These findings suggest that the hemizygosity contribution of these two genes to the NF1 microdeletion syndrome phenotype should be considered, as facial dysmorphism is a trait shared by most of the NF1 microdeletion patients.
While haploinsufficiency of genes within the deletion interval and position effect on expression regulation of genes flanking the deletion are phenomena shared by patients with the same deletion and expected to be associated to the onset of common clinical traits, the pathomechanism of the occurrence of rare clinical features of the disease remains an unresolved problem (Mautner et al. 2010). Presence of hypomorphic or recessive LoF variants is expected in human genome of healthy individuals (MacArthur et al. 2012; Deltas 2018). Their co-occurrence with the “pathogenic mutation” could aggravate the clinical phenotype as genetic modifiers and the peculiar traits in type-I NF1 microdeletion patients should be related to genetic variation. Typically, these variants belong to the same pathway or to the same interactome of the full mutation are present in the normal population with a low MAF due to negative constraints, to avoid a detrimental epistatic effect resulting from co-occurrence of more hypomorphic variants (Deltas 2018).
Because variants in more than one gene of the same pathway can contribute the severity of a specific phenotype (Löwik et al. 2008; Li et al. 2020), on the basis of the recently reported finding on a RASopathy (Ferrari et al. 2020; Tritto et al. 2023a), we investigate a possible addictive effect of RAS pathway gene variants leading to identify potential modifier genes. The targeted NGS analysis of RAS pathway and neurofibromin interactor genes allowed us to identify likely pathogenic variants within two RAS pathway genes in the following patients: N75 (RASA1) and N43 (RAF1).
The RASA1 c.2656 C > T (p.(Pro886Ser) variant was found in N75, a ten years’ old patient, with an early diagnosis (at 8 months of age), based on the presence of CALs and lentigo, who, at 4 years of age, developed a brainstem glioma that remained radiologically stable and clinically asymptomatic. His phenotype was also characterized by congenital hypothyroidism and facial hemangiomas, both of which are uncommon in NF1 phenotype. In addition, the clinical course was complicated by delayed psychomotor development and brainstem glioma diagnosed at 4 years of age. The NF1 tumor predisposing syndrome increases the risk of developing brain tumors, which are observed in approximately 15–20% of NF1 cases (Seminog and Goldacre 2013; Uusitalo et al. 2016). The optic pathway glioma, a pilocytic astrocytoma, is observed in about 15% of children with NF1, while non-optic gliomas with different histological subtypes including high-grade glioma develop more frequently at older ages (Sellmer et al. 2017). Taking into account the biological function of RASA1 and the disease related to its pathogenic variants, we propose that the severe expression of the clinical phenotype of this patient could be modulated by the p.(Pro886Ser) variant.
RASA1 variants have been associated with vascular malformation syndromes characterized by hereditary capillary malformations (CM) with or without arteriovenous malformations (AVM), arteriovenous fistulas (AVF), or Parkes Weber syndrome; however, the phenotypic spectrum of the constitutional variants of RASA1 is still being defined (Wooderchak-Donahue et al. 2018). Therefore, it is possible to hypothesize that the RASA1 variant in our patient may be responsible for the facial hemangiomas. This hypothesis is supported by the report in the ClinVar database of a patient with CM-AVM carrying the same RASA1 variant. The child also developed a brainstem glioma, which is reported in 10% of NF1 children with a median age of onset of about 7 years and an indolent course in most cases (Costa and Gutmann 2020). To our knowledge, no genotype-phenotype correlation studies have been reported in non-optic gliomas in patients with NF1. However, since RASA1 encodes an effector of the RAS pathway involved in carcinogenesis, its role in tumor etiopathogenesis in the patient cannot be excluded. In particular, the RASA1 mutation is predicted to be deleterious and involves the protein core in the RAS-GAP domain, which is responsible for the GTPase activity of the protein and for the inactivation of the RAS pathway. Mutations in additional RAS pathway genes with a similar function to neurofibromin may trigger a similar tumorigenic process leading to glioma onset. Alterations in the binding sites of either RASA1 or RAS p21 proteins are associated with basal cell carcinomas. In addition, the co-deletion of RASA1 and NF1 results in the development of T-cell acute lymphoblastic leukemia (Lubeck et al. 2015).
Likewise, a possible modulatory effect can be postulated for p.(Ser604Cys) variant in RAF1. In our cohort, the p.(Ser604Cys) variant in the RAF1 gene was found in N43, a 47 years-old patient with a common microdeletion NF1 phenotype, including ID, dysmorphic features, and cerebrovascular pathology, who was diagnosed with thyroid C-Cell Hyperplasia. The clinical picture of this patient is burdened by the developmental signs typical of NF1 microdeletion syndrome (i.e. ID and dysmorphic features) and it is possible to speculate that her features could be modulated by this variant in RAF1, which is one of the RAS pathway genes, potentially amplifying the RAS pathway dysregulation itself caused by the NF1 microdeletion. The p.(Ser604Cys) variant is a potentially damaging RAF1 mutation in the Cr3 domain, which is involved in the maintenance of RAF1 in its inactive conformation by interacting with the 14-3-3 protein family members dimers (YIP-SCHNEIDER et al. 2000). Of note, the mutation p.(Leu603Pro), adjacent to p.(Ser604Cys), observed in a Noonan patient, showed impaired kinase activity and reduced ERK activation as did a truncated RAF1 protein p.(Arg254fs) (Dhandapany et al. 2014). Pathogenic RAF1 mutations in the Cr2 domain causing Noonan syndrome are mainly associated to the onset of cardiovascular malformations, although sporadic cases of cerebrovascular abnormalities have been reported (Zarate et al. 2014; Hartill et al. 2017). The state of knowledge of its role in vasculopathy brain changes is less consolidated but there are evidences in Raf1 homozygous knock out mouse endothelial cells (Wimmer et al. 2012) of the direct critical role of Raf1 in angiogenesis. Even though full expression of microdeletion type-I phenotype and cerebrovascular abnormalities were also detected in other patients of our cohort in whom we did not find variants possibly associated with this specific clinical sign, it could be hypothesized that an effect of the p.(Ser604Cys) variant contributes to N43 patient phenotype. In this view, the occurrence of hypomorphic variants in RAS pathway genes, co-present with the main disease-mutation, could bias the clinical phenotype of microdeletion patients.
The reported genetic conditions in the above two patients and the associated variants are suggestive that peculiar phenotypes could possibly be determined by hypomorphic variants modulating the clinical picture in terms of variability and/or severity of the NF1 microdeletion syndrome. Variants in RAS pathway effectors that may function as genetic modifiers could exacerbate typical NF1 clinical features such as the neurofibroma burden or congenital abnormalities common to other RASopathies.
Regarding the pseudo-dominance effect, we identified a variant in the RNF135 gene in patient N26 who presented with learning problems, overgrowth, and coarse sections. The c.1245G > T (p.(Trp415Cys) variant in the RNF135 gene is located in the B30.2/SPRY domain. The variant is rare (MAF < 0.01) and has been previously described in three patients with Sotos syndrome features of uncertain significance (Visser et al. 2012) and reported in GnomAD also in homozygosity, thus its pathogenic significance remains to be verified.
Besides the complex effects generated by NF1 microdeletions on deregulation of multiple molecular pathways, it appears that the NF1 haploinsufficiency per se, in addition to RAS hyperactivation, directly or indirectly causes a deregulation of genes encoding neurofibromin interactors such as syndecans. It has recently been reported that syndecan SDC2, SDC3 and SDC4 were found to be hyper-expressed in both NF1 and Spinal Neurofibromatosis (SNF) patients (Bettinaglio et al. 2023). This complex molecular landscape is difficult to elucidate. The current challenge is to unravel the perturbation of homeostasis of different pathways related to neurofibromin functions, addressing the identification of possible druggable genes.
The clinical information reported in our patients lacks precise standardization, regarding the ID and the real tumor burden evaluation, because the current guidelines of Regione Lombardia do not require that all NF1 patients undergo cognitive tests or whole-body MRIs. Nevertheless, the integrated genomic approach used in this study sheds light on the heterogeneity of the pathogenetic mechanisms involved in determining the clinical characteristics and variability of this genetic disorder.
NF1 deletion syndrome is a complex disease, burdened by the variability of severe medical complications; a better knowledge of the pathogenesis underlying the clinical variability is mandatory to hypothesize future designs of personalized prevention programs for potentially life-threatening complications.

留言 (0)