
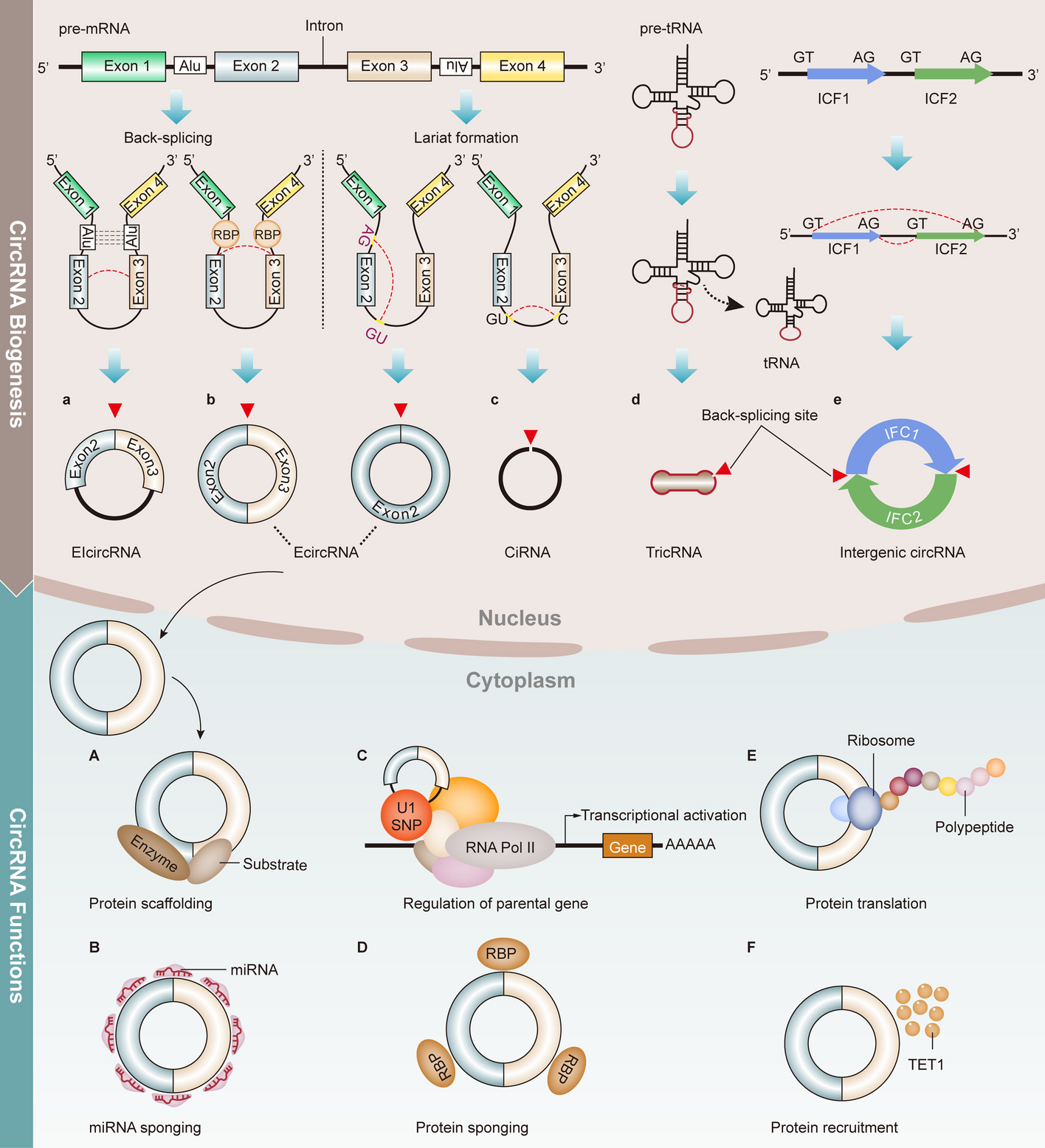
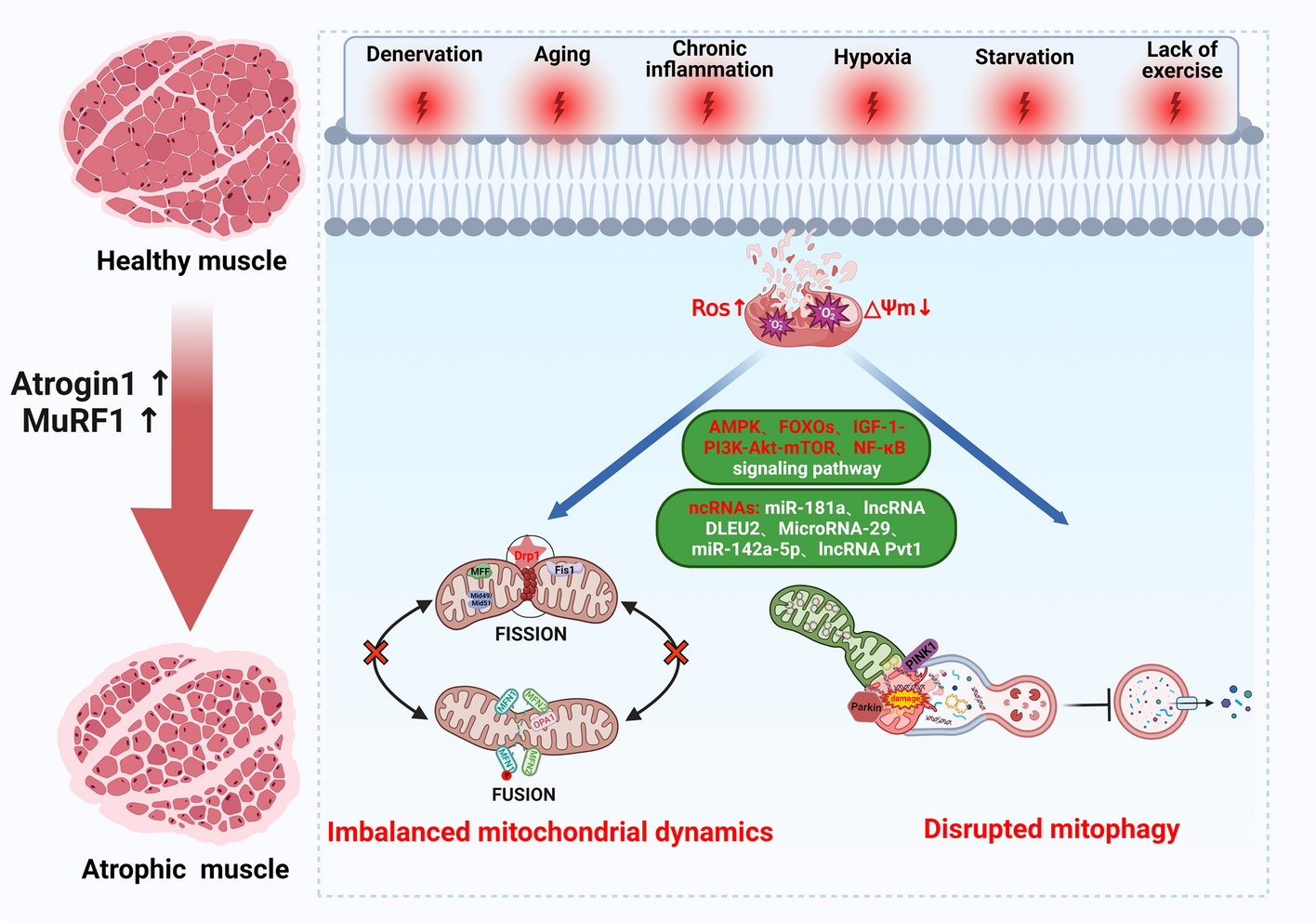
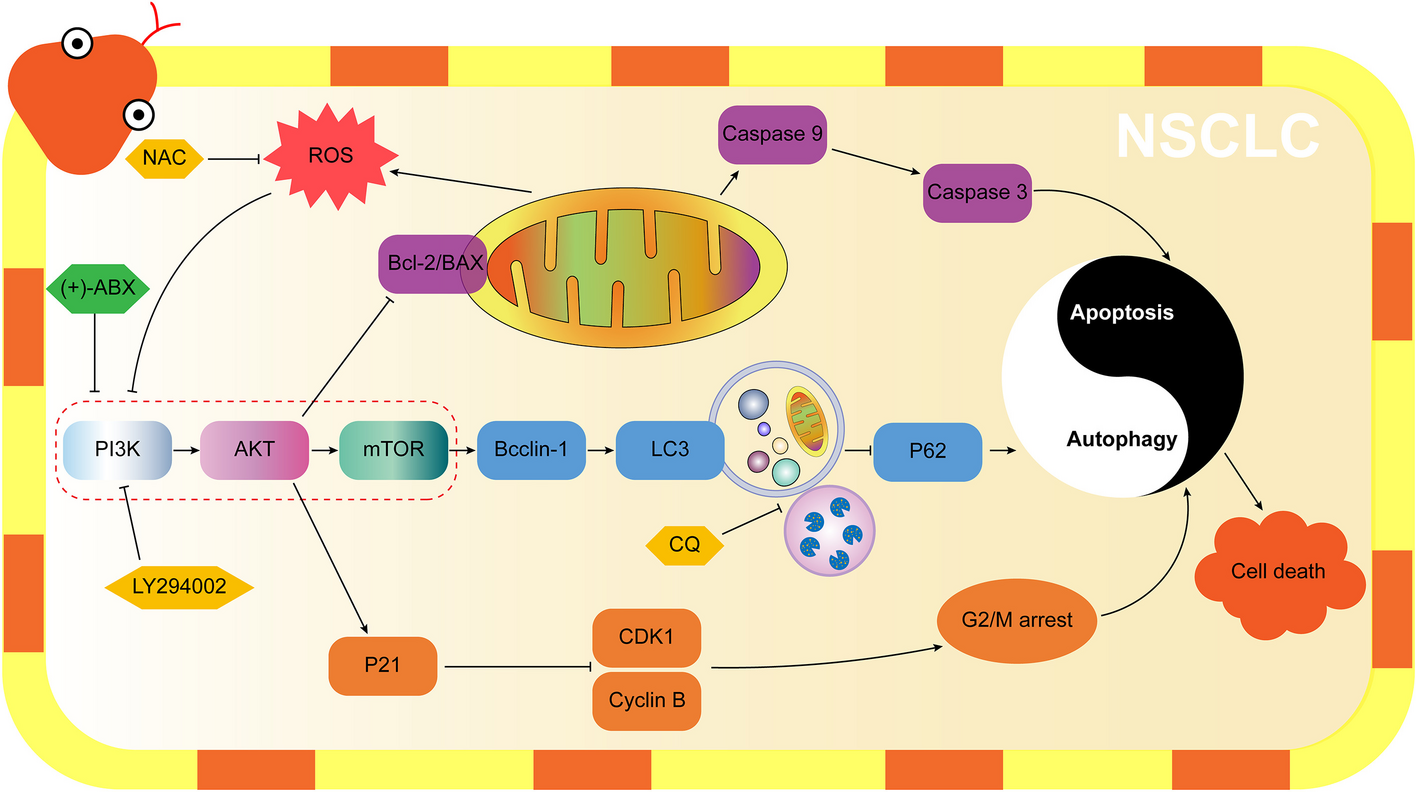
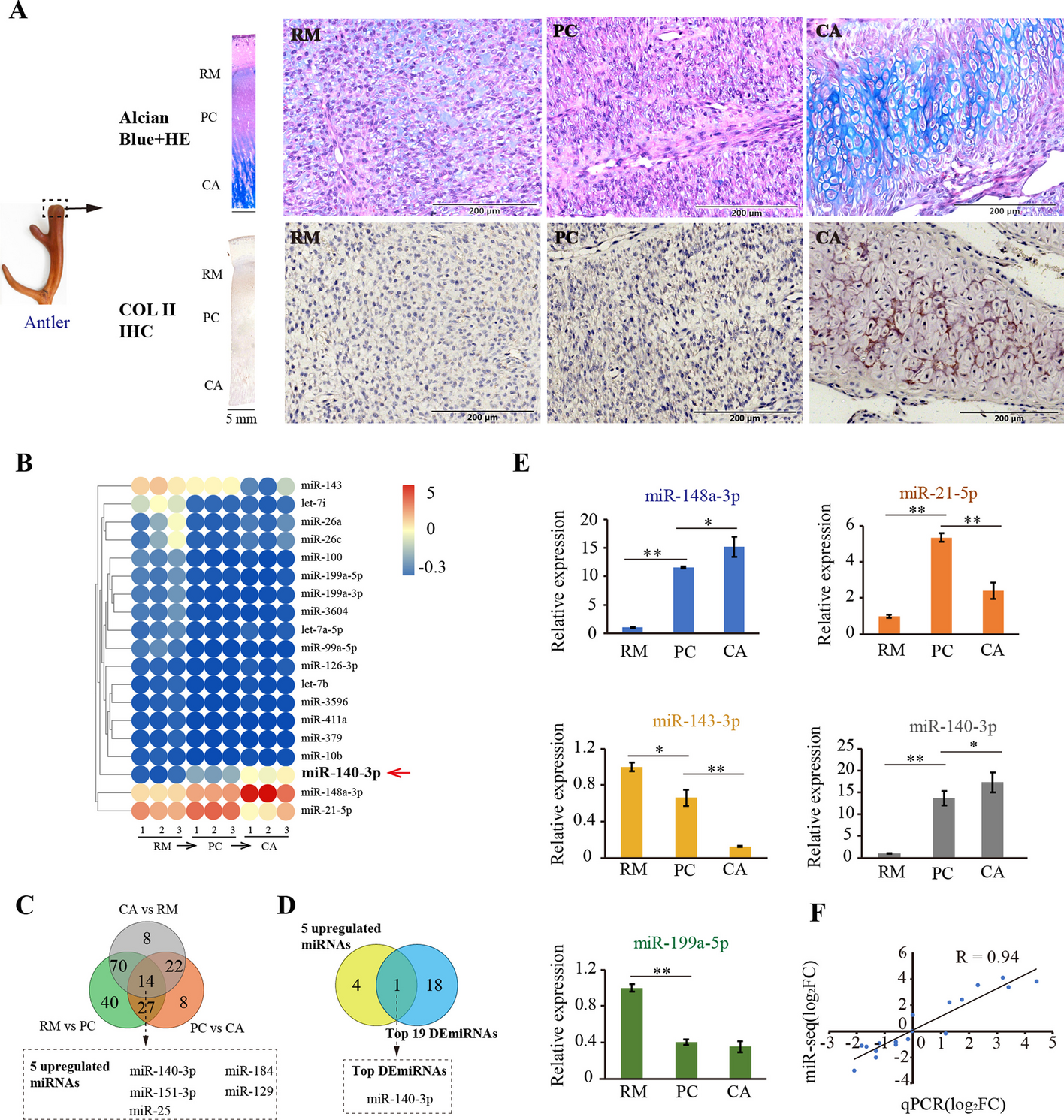
記住我


The transcriptome-wide investigation of the effects of AGC1 deficiency were performed on a differential expression design comparing three replicates of Oli-Neu cells characterized by 40% of AGC1 residual activity (kdAGC1) and three replicates of Oli-Neu (control). Western blot analysis on these cells showed a significant downregulation of the protein levels of AGC1 in samples from kdAGC1 Oli-Neu cells when compared with control (Fig. 1a). The key role of AGC1 silencing on following gene expression analysis was also confirmed through the principal component analysis (PCA) on gene expression data (Fig. 1c). We performed dimensionality reduction on these data, via PCA, which provides a convenient way to visualize the gene expression data on a two-dimensional plot, by using the values of the two first principal components, which are those explaining the largest variance in the data across all the computed components. Indeed, the first principal component identified, which explains the most variance of the data along a single axis, clearly separates the samples in control and silenced groups. Thus, the differences in gene expression that we accounted here and in the following analysis, are due to the AGC1 silencing. This certifies the good quality of the samples and the reliability of the model.
Fig. 1
Western blots and relative densitometries of AGC1 and AGC2 in Oli-Neu cells (a, b). GAPDH was used as control and for endogenous normalization. Values are mean ± standard error of the mean (s.e.m.) of at least three independent experiments; ***p < 0.001, **p < 0.01, *p < 0.05, compared with control Oli-Neu; Student’s t-test. Principal component analysis (PCA) of control and kdAGC1 samples gene expression data based on rlog-normalized reads values (c). SLC25A12 (AGC1) and SLC25A13 (AGC2) expression values [transcripts per million (TPMs)] with respective fold changes and significance levels (d, e)
To assess the levels of inhibition of AGC1 at RNA level, we compared both AGC1 and AGC2 quantified levels of transcripts [transcripts per million (TPMs)] in kdAGC1 and control samples, observing a similar degree of downregulation for both genes: mean TPM values for AGC1 were respectively 26.9 for control and 20.3 for kdAGC1, with a significant log2(fold change) (LFC) of −0.39 (adjusted p = 0.039) (Fig. 1d). Mean TPMs for AGC2 were respectively 1.06 for control and 0.429 for kdAGC1, with a significant LFC of −0.97 (adjusted p = 0.027) (Fig. 1e). However, from western blot analysis on AGC2 protein content, an opposite result was obtained, with an upregulation of the second isoform of the carrier in silenced cells respect to control ones (Fig. 1b). This suggests a possible compensatory mechanism that is induced to counteract the silencing of AGC1 and that could explain the lack of mitochondrial activity dysfunction in AGC1 partially silenced Oli-Neu cells [11].
When estimating LFCs, it is known that genes with low counts or a high coefficient of variation can be a problem, especially in experimental setups where the number of replicates is very small, leading to LFCs value that are often not reflected by the true expression levels in the biological model under study [25]. Several methods have been proposed to mitigate this problem, and we chose to use apeglm [26] because of its many advantages among some of which are not needing to set fixed thresholds for filtering low counts genes, the use of pseudocounts and its easy implementation in our analysis pipeline. By visualizing the effect size against each gene’s counts (MA plot), it is clear how the use of this shrinkage significantly lowers the effect size of those genes with lower counts, while no impact at all is shown for those genes with a higher gene count (Additional file 5: Fig. S1).
Looking at the genes significantly altered (Fig. 2a), we observe 769 significantly altered genes, divided in 356 downregulated genes and 413 upregulated genes, with a prevalence of downregulated genes among the most significant ones, such as CHODL (also confirmed via real-time PCR, Fig. 3g), encoding for the transmembrane protein Chondrolectin, and MRPL55, encoding for a mitochondrial ribosomal protein. Among the genes with the strongest kdAGC1-induced upregulation we identify several ones encoding for cell adhesion molecules, such as CORO2A, ITGA5, BGN, and FAT3, suggesting that that both cell–cell adhesion and the interactions with the extracellular matrix might be altered (Additional file 5: Fig. S2).
Fig. 2
Volcano plot of kdAGC1 samples versus control (a). The X axis represents the magnitude of the change in expression levels, while the Y axis represents the significance of the change. Significance threshold was set to p adjusted < 0.05. Points in red are genes that are both significant and show |log2FC| > 1, while blue points indicate genes that are significant and have |log2FC| < 1. Points in gray represent genes that do not pass both thresholds. Enrichment score values (“combined score”) and significance levels for the top ten scoring Gene Ontology pathways obtained from gene set enrichment analysis (GSEA) (b). Scaled TPM values heatmap of a panel of genes that resulted as significant form differential gene expression analysis, for which log2FC and p adjusted values are reported on the right of the heatmap (c)
Fig. 3
Western blot and relative densitometries of FASN (a, b), ACSS1 (a, c), and precursor and cleaved SREBP1 (a, d, e) expression in Oli-Neu cells; GAPDH was used for endogenous normalization. Confocal microscopy images (f) in Oli-Neu cells; nuclei were labelled with DAPI. Scale bar, 20 µm; 100× objective. Reverse transcription real-time PCR analysis (g). GAPDH were used as endogenous controls. Values are mean ± standard deviation (SD) of at least three independent experiments; ***p < 0.001, **p < 0.01, *P < 0.05, compared with control Oli-Neu; Student’s t-test
To more concisely determine which cellular pathways were significantly altered we conducted a pathway enrichment analysis based on the Molecular Signature Database (MSigDb), restricting the analysis to pathways from the Hallmark, Curated, and Ontology gene sets. The results from the analysis were further filtered to unify similar pathways, due to the redundancy that is found in some of the gene sets used for the analysis, especially the Curated and Ontology gene sets. This analysis further proves the impact that the downregulation on AGC1 has on the capability of the cell to bind to other cells and the extracellular matrix (Fig. 2b). Several pathways, including those related to biological adhesion, and locomotion (intended as cell motion) are significantly upregulated in kdAGC1 samples. In addition, the neurogenesis pathway is also upregulated in kdAGC1, a result that is also supported by recent studies on neurospheres derived from heterozygous mice, where a decrease in terms of number of cells for the OPCs population, in favor of an increase in mature OLs, neuronal progenitors, and astrocytes is reported [11]. This could be explained by the fact that NG2+ progenitor cells can display a multipotent phenotype in vitro and generate electrically excitable neurons, as well as astrocytes and oligodendrocytes, reflecting an intrinsic property, rather than reprogramming [34]. Therefore, in AGC1 deficiency, where OPCs are induced to undergo differentiation at the expense of proliferation, together with oligodendrocytes markers’, also astrocyte, neuron, and Schwann markers’, expression could be upregulated, probably due to their neurogenic potential. Even though we are not able to see a significant downregulation on a common proliferation marker such as Ki67 [35], and we did not see a specific downregulation in glial cells proliferation pathways, we observed an upregulation in astrocytes, mature neurons, and Schwann cells markers, with no significant results for OPCs markers, meaning that a lack of AGC1 could drive the differentiation of the affected cells.
One of the key pathways in which AGC1 is involved through its activity in MAS is the complete glucose oxidation, by sustaining the entry of the glycolysis-derived pyruvate in the mitochondria and subsequent oxidative decarboxylation of this metabolite via tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) [5]. In neurons, the acetyl-CoA produced from pyruvate in the mitochondria can also be exported to the cytosol in the form of citrate, making available acetyl groups in this compartment through the citrate lyase. Cytosolic acetyl-CoA are then transferred to OLs as N-acetyl aspartate (NAA), thus entering the lipid biosynthetic pathway essential for the myelination and re-myelination processes of these cells [6].
To observe alterations in genes involved in myelination in silenced versus control samples, we selected all genes that belonged to a Gene Ontology myelin-related pathway that resulted significant from differential expression analysis (Fig. 2c). From this analysis, fatty acids synthase N (FASN), which regulates the fatty acids synthesis via production of palmitate from acetyl-CoA and malonyl-CoA [36], as well as myelination and remyelination processes, and sterol regulatory element binding protein 1 (SREBP1), a master regulator of fatty acids synthesis, which controls the expression of many enzymes involved in this pathway (as FASN) [37, 38], come out to be downregulated. Interestingly, AKT1 is likewise downregulated, suggesting a possible role of this protein and associated metabolic pathway as responsible for SREBP1-altered maturation and downregulation, since it controls translation of many genes involved in lipogenesis [39].
With an opposite trend, PTN and PTPRZ1 are found to be upregulated in kdAGC1 samples. According to recent studies, PTPRZ1 plays roles in cell proliferation, cell adhesion, migration, cancer stem cells, and treatment resistance through its interaction with various molecules [40, 41]. Additionally, other many genes encoding for markers of mature OLs resulted in upregulation in RNA-seq analysis, all controlling the correct temporal OPCs proliferation and differentiation process [42, 43]. Among these, myelin transcription factor 1 (MYT1), which regulates a critical transition in oligodendrocyte lineage cell development by modulating OPCs proliferation relative to terminal differentiation together with upregulation of myelin gene transcription, and oligodendrocyte myelin glycoprotein (OMG), involved in the formation and maintenance of myelin sheaths, are crucial in brain development and regeneration after injury [44].
It is interesting to point out an upregulation in retinoic acid (RA) receptors gene expression (RARG and RARB) in silenced samples (Additional file 1); RA is known to be involved in the transition from NSCs to OPCs [45], but there are conflicting opinions on whether it inhibits myelination [46] or stimulates it.
An interactive website has also been created [27], which allows for an easy and interactive exploration of the dataset, allowing to explore differential expression analysis results, which are reported as a table where genes can be filtered to selectively show in the volcano plot and the boxplots reporting count values. Search bars have also been added to navigate the dataset based on specific genes, groups of genes reported in Gene Ontology Biological Process (GOBP), Kyoto Enclyclopedia of Genes and Genomes (KEGG), and Reactome pathways, or based on log2FC and adjusted p values.
Experimental validation on Oli-Neu cellsTo validate the RNA-seq results of the model of AGC1 deficiency in Oli-Neu cells, we performed real-time PCR analysis on a selection of the genes that were found to be significantly altered in their expression levels. This analysis confirms the RNA-seq results for all the tested genes, supporting the validity of transcriptomics results (Fig. 3g).
Subsequently, since from RNA-seq data a large part of altered genes belongs to the fatty acids and myelin lipids synthesis pathway (Fig. 2c) and because of the fundamental role of these pathways in OPCs, more detailed analyses (real-time PCR, western blot and immunofluorescence) were performed on both control and kdAGC1 Oli-Neu cells.
For this purpose, we chose three proteins, mostly involved in fatty acids and acetyl-CoA synthesis: SREBP1, FASN, and acetyl-CoA synthetase 1 (ACSS1), which converts acetate into acetyl-CoA [47]. Both these two last enzymes are activated by SREBP1, upon decrease of sterol levels [47, 48].
The real time PCR shows a significant downregulation of SREBP1 (Fig. 3g), in line with RNAseq data, confirmed also with western blot analysis (Fig. 3a, d), which shows a reduced protein content for both the inactive/precursor and active/cleaved form of this protein. Even though SREBP1 is downregulated, we observed an upregulation of ACSS1 transcript (Fig. 3g), in the silenced Oli-Neu cells. However, at the protein level in western blot analysis, this enzyme does not show a similar increment (Fig. 3a, c). This could be explained by the fact that ACSS1 is only partially controlled by SREBP1 activity [49]. We can hypothesize that this protein is normally synthesized since the cell preferentially obtains acetyl-CoA from citrate by ATP citrate lyase (ACLY) in physiological conditions, and only in case of nutrients deprivation relies on this pathway [50].
Interestingly, in kdAGC1 Oli-Neu cells, although there is a strong background noise for this specific antibody, FASN came out significantly downregulated at the protein level, considering both the single 240 kDa band and the total bands (Fig. 3a, b). This could be due to the reduced activity of SREBP1, as well as due to a post-translation dysregulated process of acetylation/deacetylation that could destabilize/stabilize FASN protein. Indeed, deacetylation can protect the enzyme from degradation, but, as previously seen [12], HDAC3—the enzyme responsible for this modification—is downregulated in silenced cells, probably causing the rapid turnover of this protein, upon acetylation [51]. Generally, no altered subcellular localization was detected for these candidate proteins in kdAGC1 Oli-Neu cells compared with control, through immunofluorescence analysis (Fig. 3f).
In parallel with these proteins, to study fatty acids synthesis pathway, other transcripts were analyzed, revealing alterations in other main enzymes and transcription factors involved (Fig. 3g). An example is SREBP2, the isoform of SREBP1, which is mainly responsible of cholesterol pathway [37]. This suggests that not only fatty acids synthesis, but also cholesterol production could be impaired in silenced Oli-Neu cells, hampering the formation of new cell membranes, essential for myelination [52]. Taken together, these results suggest that AGC1 silencing can compromise the fatty acids synthesis pathway, including principal transcription factors and enzymes, as SREBP1 and FASN.
Experimental validation on neurospheresA similar scenario to what we observed in the experimental analysis of Oli-Neu cells is also present in the second AGC1 deficiency in vitro model, constituted by neurospheres obtained from 8-month-old mice’s subventricular zone (SVZ). This in vitro model primarily consists of neurons, astrocytes, and OLs progenitors’ pool. The three-dimensional model more accurately replicates the conditions observed in patients with AGC1 deficiency, as homologous recombination of AGC1 mRNA enables the insertion of a premature stop codon in exon 2 and 3, leading to the creation of heterozygous mice. These retain a 50% of residual carrier activity, which mimic the pathological conditions found in patients [11]. For completeness, we first performed on this model the real-time PCR on the same panel of genes, previously chosen for RNA-seq data validation in Oli-Neu cells. In general, out of 13 genes, 9 have consistent results with Oli-Neu kdAGC1 at the transcripts level (Fig. 4), possibly due to neurospheres being constituted by a mixed pool of NSCs. This means that each sphere can give rise to all the three different neural cell types, which contributes differently to the enzymatic and protein set. In general, we can state that this analysis confirms the reliability of both models of AGC1 deficiency and their similarities, above their intrinsic differences. Only CHODL represents an exception because for both wild-type and heterozygous neurospheres, the mRNA levels are low and seem to be undetectable by the real-time PCR, in contrast with Oli-Neu results.
Fig. 4
Western blot and relative densitometries of FASN (a, b), ACSS1 (a, c), and precursor and cleaved SREBP1 (a, d, e) expression in AGC1+/− and AGC1+/+ neurospheres; GAPDH was used for endogenous normalization. Confocal microscopy images (f) in neurospheres; nuclei were labeled with DAPI. Scale bar 50 µm; 60 × objective. Reverse transcription real-time PCR analysis (g). GAPDH were used as endogenous controls. Values are mean ± standard deviation (SD) of at least three independent experiments; ***p < 0.001, **p < 0.01, *p < 0.05, compared with AGC1+/+ control neurospheres Student’s t-test
Along with the experiments we previously carried out for Oli-Neu cells, we performed western blot and immunofluorescence analysis, to better characterize alteration upon major players of the fatty acids and myelin lipid synthesis pathway in neurospheres. In accordance with real-time PCR and results in Oli-Neu cells, SREBP1 shows a reduction of 40–50% even at the protein level (Fig. 4a), in heterozygous neurospheres compared to wild-type ones, suggesting an altered control of fatty acids synthesis. In contrast, FASN exhibits an upregulation both at mRNA and protein level in AGC1+/− neurospheres (Fig. 4a). Even though, in this latter analysis, the FASN antibody had shown a strong background noise, upregulation was confirmed both considering the single 240 kDa band and the total bands. This discrepancy with the Oli-Neu cells can be explained by the heterogeneous pool of the neurospheres and the cell-specific enzymes present in this model. Moreover, HDAC3 is upregulated in heterozygous spheres [12], so that it can protect FASN from degradation. Similarly, ACSS1 is upregulated in AGC1+/− neurospheres. Here, probably, the contribution of neuronal and astrocyte progenitors—toward which the heterozygous neurospheres tend to differentiate more easily [10]—can explain the upregulation of this enzyme and the differences with Oli-Neu cells. Also, for this in vitro model, the subcellular localization of these proteins does not seem to change in heterozygous neurospheres, in immunofluorescent images (Fig. 4f).
As in Oli-Neu cells, SREBP2, the second isoform of SREBP1, shows a downregulation in heterozygous neurospheres (Fig. 4g), further suggesting a possible dysregulation of cholesterol synthesis. These experiments confirm an alteration at the level of fatty acids, acetyl-CoA and myelin lipids synthesis pathway, which play a crucial role in cell proliferation, differentiation and function, not only in the Oli-Neu cell models, but also in neurospheres.
Master regulator analysis shows upregulation in Prox1 and downregulation in Smarcc2Understanding the full effects of AGC1 deficiency at the molecular level demands further investigation of the transcriptional data, both in terms of differential splicing and via master regulator analysis, whose purpose is to detect the most likely transcription factors in charge of the observed transcriptional changes [53].
Master regulator analysis was performed using the corto R package [32], which is a combined extension of the well-established algorithms for gene network generation ARACNe-AP [54] and interrogation VIPER [53]. The idea behind master regulator analysis is to infer the kdAGC1-specific activation levels (represented by enrichment scores) of histologically specific and accurate gene co-expression networks centered around transcriptional regulators. To do so, a network of gene interactions was computed from a large (> 100 samples) tissue-specific RNA-seq dataset to establish regulons, which are groups of target genes that are controlled by a common master regulator. The activation level of a master regulator is then inferred on the basis of the expression levels of its target genes in a specific experimental setup (e.g., if all the targets of Myc, which is considered to be a master regulator, are positively upregulated in a treated versus control experiment, then the activity of Myc will be positive and very high. Different degrees of activation are determined by the differential expression levels from the results of an RNA-seq analysis).
The main challenge for these types of algorithms is to find an RNA-seq dataset that closely resembles the tissue or the cell type on which the experiment has been conducted, since it requires to have a large enough number of samples to accurately infer the gene networks. To do so, we tested two different networks obtained from GTEx tissues: frontal cortex and hippocampus [55], converted to mouse orthologs through the DIOPT consensus phylogenetic approach [56].
This analysis allowed us to identify two master regulators, PROX1 and SMARCC2, that we hypothesize to be key factors at the basis of the transcriptional alterations that we showed earlier. One of the most relevant master regulators whose regulon is found to be consistently activated in both networks is PROX1. It is a homeobox transcription regulator that has been shown to work a switch determining cell-fate decisions between neurogenesis and oligodendrogenesis, acting as a strong repressor for neuronal lineage commitment of NSCs deriving from subventricular zone [57]. However, it not only regulates cell fate at early stages, but it is highly expressed both in OPCs and OL and specifically its expression increases along with OPCs differentiation progression [58]; whereas it reduces the OPCs proliferation through inhibition of NG2, a proteoglycan involved oligodendrocyte renewal and maintenance [59, 60]. All these data are in line with our in vitro AGC1 deficiency model, which show reduced proliferation of OPCs and reduced NG2 expression together with premature differentiation [11, 12].
SMARCC2 is a master regulator, whose regulon we found to be significantly and negatively enriched. Human SMARCC2 encodes for BAF170, a core subunit of the ATP-dependent BAF complexes (the mammalian ortholog of the SWI/SNF complex) that are known to regulate chromatin remodeling and gene expression during embryogenesis and play a crucial role in neurodevelopment. Various studies on SMARCC2 knockout mice showed that its absence brought to a degradation of BAF complexes causing an impairment of fore brain development [61]. More specifically, BAF170, along with BAF155, is essential for oligodendrogenesis and its deletion impairs proliferative process—depleting the number of PDGFRα positive cells in mice forebrain—and differentiation from OPCs towards immature OLs [62]. These data are in line with those previously found [11] regarding defects of this AGC1 deficiency model on proliferation and differentiation process and their tight link with epigenetic dysregulation [12], reinforcing the already known bond between epigenetic mechanisms and OLs maturation [63]. An extended analysis including more identified master regulators at lower significance is reported in Additional file 5: Fig. S4.
Splicing analysisTranscript splicing has been known to play a role in diverse aspects that affect oligodendrocytic cells, especially when it comes to myelination: most of the proteins that are found in myelin sheaths are reported to undergo mRNA splicing before being translated, with splicing isoform expression that can vary based on the stage of the myelination process [64,65,66].
To study the differential expression of splicing isoforms in our kdAGC1 versus control samples we used MAJIQ [33], a computational framework able to identify de novo transcript isoforms and the associated classical splicing variations (e.g., exon skipping and exon inclusion) and non-classical splicing variations (such as intron retention, alternative donor site and alternative acceptor site). Results are reported as a difference of percent spliced in (PSI), where PSI is the relative ratio of isoforms including a specific splicing junction or retained intron, and their difference (dPSI) measures the change of the ratios between two conditions of each splicing event, with a value that can range from −1 to 1.
Alternative splicing analysis allowed to identify significant effects of AGC1 silencing on the relative isoform abundance of a myelin-related protein, PMP22 (Fig. 5b), which is known to be mostly expressed in myelinating cells of the peripheral nervous system but has also been detected in the CNS. PMP22 is required for the correct myelination of peripheral nerves and in keeping axon myelinated [67]. Mutations or genetic alterations, such as gene duplication, in PMP22 are responsible for different inherited peripheral neuropathies, among which we find Charcot–Marie–Tooth type 1A (CMT1A) hereditary neuropathy with liability to pressure palsies (HNPP) and a subtype of Dejerine–Sottas Syndrome (DSS) [68]. In kdAGC1 samples we see a specific local splicing variation (in blue, Fig. 5b) of exon 5 splicing upstream with exon 3 that is found 50% more often compared with control samples, while control samples do not look like they have such large changes in local splicing variations compared with kdAGC1 ones. A case of two siblings with CMT1 showed a deletion in PMP22 exon 4 has recently been reported [69], showing how the deletion of this exon causes a segregation of PMP22 to the endoplasmic reticulum instead of localizing the protein to the plasma membrane, contributing to the pathogenesis of CMT1 (Fig. 5a).
Fig. 5
Master regulator analysis of PROX1 and SMARCC2 in two different networks (frontal cortex and hippocampus) showing the most significant transcription factors with differentially activated networks in kdAGC1 versus control (a). The upper bar shows the symbol of the tested master regulator, its normalized enrichment score (NES), with its cell colored in red for activated regulons and blue for downregulated regulons, and the associated adjusted p value. The barcode graph indicates the distribution of activated (red bars) and repressed (blue bars) targets of a master regulator. Target genes are ranked from left to right from most downregulated to most upregulated
留言 (0)