
記住我

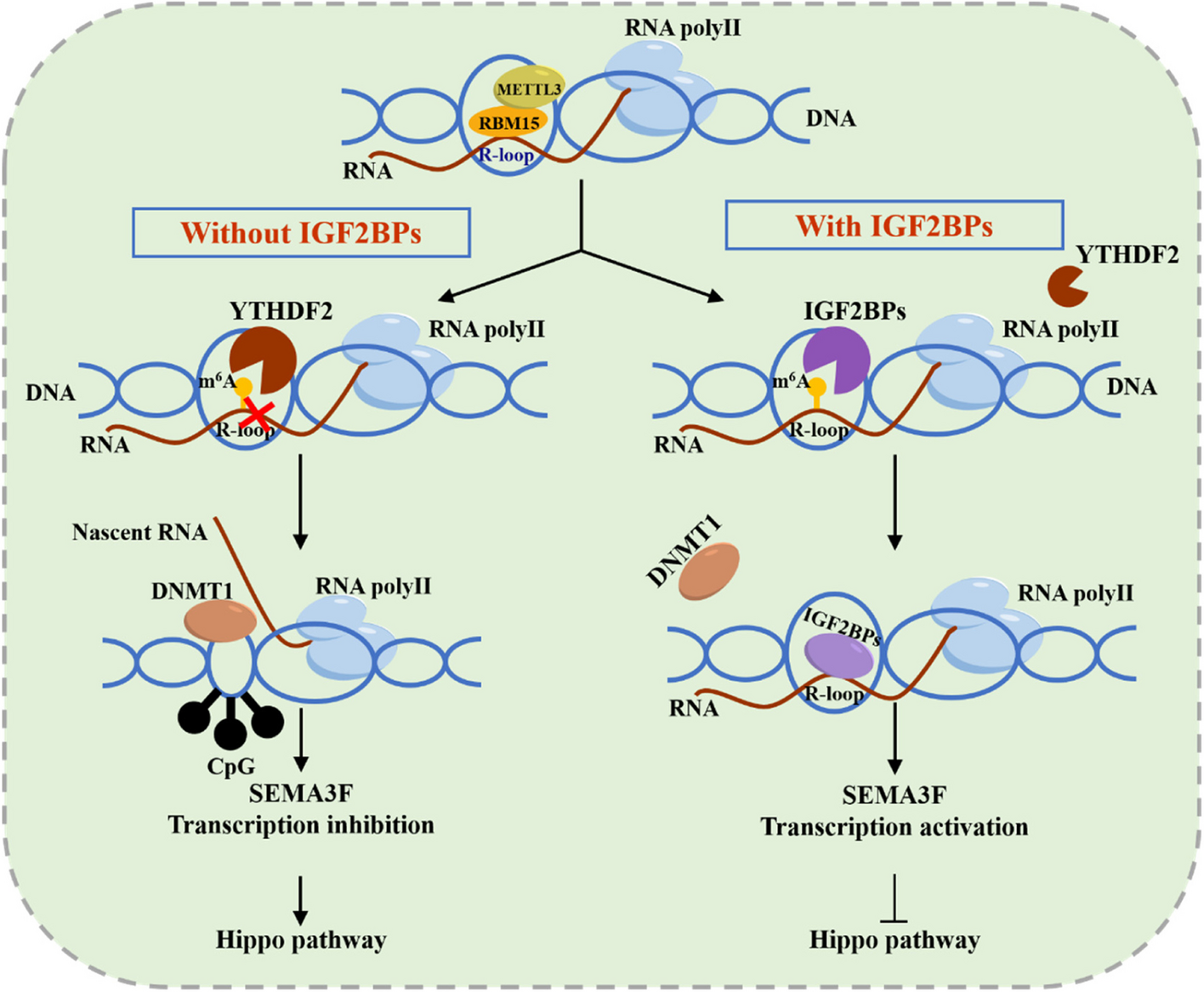

GO, the pioneering ADC drug developed by Pfizer, holds the distinction of being the first ADC to receive global market approval. Comprising a humanized mAb targeting CD33 and a cytotoxic N-acetyl-γ-calicheamicin connected via a cleavable hydrazone linker, GO operates on a therapeutic principle designed for patients with AML [8]. The mechanism involves GO binding to the CD33 antigen, forming the GO–CD33 complex, which is then internalized into AML primary cells [9]. Following positive outcomes from three early clinical trials, the FDA granted approval of GO in 2000, specifically for the treatment of patients with CD33-positive AML aged over 60 who were ineligible for cytotoxic chemotherapy [10].
However, safety concerns surfaced during the Southwest Oncology Group (SWOG) S0106 study, designed to assess the efficacy of GO across all cytogenetic risk groups in adult patients below 60 years of age with AML. The study revealed a higher fatal induction toxicity rate in the GO + cytarabinealone group compared to the cytarabinealone group (5.5% vs. 1.4%) [11]. Consequently, Pfizer withdrew the product from the market in June 2010. The safety and therapeutic efficacy of GO were reevaluated at a lower dose (3 mg/m²) in combination with chemotherapy in the Acute Leukemia French Association (ALFA)-0701 phase III clinical trial. The study indicated varying incidences of grade ≥ 3 adverse events (AEs) in the GO combined with chemotherapy and chemotherapy alone groups, including veno-occlusive liver disease (2% vs. 0%), hemorrhage (18% vs. 9%), and infections (47% vs. 39%), respectively [12].
Additional trials, namely MyloFrance-1 and AML-19, were conducted to assess the safety and efficacy of GO [4, 13]. The MyloFrance-1 study demonstrated significant toxicities associated with GO treatment, such as myelosuppression, infusion reactions, infections, bleeding, and hepatotoxicity. However, the data suggested that the anticipated clinical benefits for patients with CD33-positive relapsed/refractory AML outweighed safety concerns when treated with 3 mg/m² GO on days 1, 4, and 7 [4]. In the AML-19 study focusing on overall survival (OS), results indicated a promising improvement in OS for elderly patients with AML unsuitable for intensive chemotherapy compared to best supportive care. The toxicity was manageable, with no additional adverse effects observed [13]. Based on these studies, the FDA granted approval of GO in 2017 [14]. Subsequently, in pediatric AML, the Children’s Oncology Group’s AAML0531 trial demonstrated improved prognosis for pediatric patients treated with GO [15].
The final efficacy and safety update from the open-label, phase III ALFA-0701 trial revealed that the addition of GO to standard chemotherapy significantly extended event-free survival (EFS) in patients with newly diagnosed de novo AML [16]. In a randomized, open-label, multicenter phase III trial (AMLSG 09–09), the primary investigation focused on the efficacy of intensive chemotherapy with or without GO in patients with NPM1 mutant AML. The results demonstrated a significant reduction in the cumulative relapse rate when GO was combined with chemotherapy. AEs (grade ≥ 3) and their incidence in the GO combined with chemotherapy and chemotherapy alone groups included febrile neutropenia (47% vs. 41%), thrombocytopenia (90% vs. 90%), pneumonia (25% vs. 22%), and sepsis (29% vs. 25%), respectively [17].
A retrospective analysis gathered data on 35 children with refractory or relapsed AML treated with GO in Poland from 2008 to 2022. Outcomes indicated that 18 children achieved complete response (CR), 14 did not respond to treatment, and 3 progressed. Among the 18 children with CR after GO treatment, allogeneic hematopoietic stem cell transplantation was performed. The 5-year OS for the entire cohort post-GO treatment was 37.1% ± 8.7%. Patients with strong CD33 expression (more than 50% positive cells) demonstrated a trend towards better outcomes compared to those with low CD33 expression. Common AEs included bone marrow aplasia, unexplained fever, infections, and elevated liver enzymes [18].
In the UK NCRI AML18 trial, investigators explored the benefits of fractionated versus single-dose GO in elderly patients with AML. Results indicated that a fractionated regimen was more effective than a single dose in clearing leukemia in older individuals without adverse genetic risk [19]. In a phase IV study evaluating the QT interval, pharmacokinetics, and safety after fractionated GO administration in patients with relapsed/refractory CD33-positive AML, findings suggested that a fractionated GO dosing regimen did not pose a clinically significant safety risk for QT interval prolongation. Treatment-emergent adverse events (TEAEs) were consistent with the previously reported safety profile of GO [20].
Evidence has shown that GO, when combined with standard induction chemotherapy, enhances the prognosis for newly diagnosed intermediate cytogenetic risk AML [21]. The use of GO in combination with fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin has demonstrated improved EFS in young patients newly diagnosed with AML, and enhanced OS in patients with NPM1 and FLT3 mutations [22]. Collectively, these findings suggest that GO, whether administered as a standalone agent or in combination, slows disease progression and is deemd safe, efficacious, and feasible in patients with CD33-positive AML at their initial diagnosis.
Brentuximab vedotin (BV)BV, initially developed by Seagen (formerly Seattle Genetics) and later co-developed with Takeda, stands as the second approved ADC drug. It comprises brentuximab, a chimeric IgG1 mAb targeting CD30, a maleimide linker moiety (a cleavable dipeptide linker, mc–VC–PABC), and monomethyl auristatin E (MMAE). BV specifically targets the CD30 antigen expressed in Hodgkin lymphoma (HL) and anaplastic large cell lymphoma (ALCL) [23].
In a phase I study evaluating the efficacy and safety of BV for the treatment of HL and ALCL, 45 patients with relapsed/refractory CD30-positive hematological malignancies received BV at doses ranging from 0.1 to 3.6 mg/kg of body weight every 3 weeks. Results indicated objective response in 50% of cases, with a median duration of response (DOR) lasting at least 9.7 months. Most AEs were of grade 1 and 2 severity, with the common ones including fatigue, fever, diarrhea, nausea, neutropenia, among others [24]. In a phase II trial, BV demonstrated effectiveness in 75% and 87% of patients with HL (102 patients) and ALCL (30 patients), respectively [25].
A phase III study exploring BV in the treatment of cutaneous T-cell lymphomas revealed significant improvement in mycosis fungoides or primary cutaneous ALCL. Moreover, BV demonstrated the ability to alleviate itch and pain caused by lymphoma without negatively impacting the patients’ quality of life (QoL) [26]. In 2018, the FDA approved BV in combination with CHP (i.e., cyclophosphamide, doxorubicin, and prednisone) for treating adult patients with previously untreated systemic ALCL or other CD30-expressing peripheral T-cell lymphomas (PTCL), including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified [27].
In a multicenter real-world study conducted between 2020 and 2022, researchers enrolled 104 patients with lymphoma receiving BV for the first time. The results demonstrated an objective response rate (ORR) of 64.5%, with 6-month progression-free survival (PFS) and OS rates reaching 77.2% and 90.1%, respectively. The 12-month PFS and OS rates were reported at 77.2% and 79.9%, respectively. The most prevalent AEs were hematological disorders, particularly neutropenia [28].
In an open-label, single-arm, multicenter phase I/II trial, 41 patients with HIV-related HL received BV in combination with doxorubicin, vinblastine, and dacarbazine. Results indicated that all 37 patients who completed treatment achieved CR. The 2-year PFS was 87%, and the OS rate was 92%. The most common grade 3 or worse AEs included peripheral sensory neuropathy (10%), neutropenia (44%), and febrile neutropenia (12%) [29].
Trastuzumab emtansine (T-DM1)T-DM1 is an ADC drug formed by linking the HER2-targeting drug trastuzumab with emtansine (also known as DM1) via a thioether linker. The targeting action of trastuzumab selectively transports the highly active cytotoxic small molecule drug DM1 into tumor cells with HER2 overexpression, releasing the drug through endocytosis. This mechanism not only significantly reduces toxicity and side effects but also enhances the targeting role [30].
The international multicenter phase III clinical trial, EMILIA, conducted by Verma et al., affirmed the clinical role of T-DM1 in HER2-positive advanced breast cancer. In patients with HER2-positive metastases or advanced breast cancer treated with trastuzumab and paclitaxel, T-DM1 demonstrated enhanced treatment efficacy, a higher safety profile, and fewer adverse effects [31]. In the TDM4450g study, T-DM1 showed generally favorable tolerance in patients with HER2-positive metastatic breast cancer. TEAEs with an incidence rate of > 40% included fatigue (49.3%), nausea (49.3%), an increase in serum aspartate aminotransferase (43.5%), pyrexia (40.6%), and headache (40.6%) in the T-DM1 group [32].
The TH3RESA study validated the effectiveness of T-DM1 in breast cancer patients who progressed after second-line and above treatment. Results indicated a significantly improved median PFS in the T-DM1 group, along with a prolonged median OS and a lower proportion of ≥ 3 adverse reactions compared to the control group [33]. A real-world study presented at the 2019 European Society for Medical Oncology (ESMO) congress from a US database confirmed the benefit of T-DM1 in patients who had failed dual-target therapy with trastuzumab and pertuzumab [34]. The NCCN Breast Cancer Guidelines designate T-DM1 as the preferred second-line treatment for HER2-positive advanced breast cancer [35].
In adjuvant therapy for residual invasive HER2-positive early breast cancer, T-DM1 plays a crucial role. The KATHERINE study indicated that the T-DM1 group exhibited an improved 3-year disease-free survival (DFS) rate and a significantly reduced risk of recurrence or death. This study establishes T-DM1 as the new standard treatment for patients with residual lesions after neoadjuvant therapy for HER2-positive breast cancer [36].
In the ATEMPT trial, the objective was to assess whether T-DM1 treatment resulted in lower toxicity compared to paclitaxel plus trastuzumab, while still achieving clinically acceptable invasive DFS in patients with stage I HER2-positive breast cancer. The study revealed that the 3-year invasive DFS for T-DM1 reached 97.8%, and patients treated with T-DM1 experienced less neuropathy and alopecia than those treated with paclitaxel plus trastuzumab [37].
In the WSG-ADAPT-TP phase II trial involving 375 hormone receptor-positive or HER2-positive patients, randomization into three groups (T-DM1, T-DM1 + endocrine therapy, trastuzumab + endocrine therapy) resulted in similar 5-year invasive DFS rates (88.9%, 85.3%, and 84.6%, respectively) and OS rates (97.2%, 96.4%, and 96.3%, respectively) [38].
A phase I trial enrolled 12 patients with HER2-positive breast cancer and brain metastases, investigating the combination of T-DM1 and metronomic temozolomide. The study indicated low-grade toxicity and potential activity in the secondary prevention of HER2-positive brain metastases. Grade 3 or 4 AEs included thrombocytopenia, neutropenia, lymphopenia, and CD4 reduction [39].
Additionally, in the phase II KAMELEON study (NCT02999672), the aim was to explore tumor HER2 expression and its impact on T-DM1 response in patients with HER2-positive urothelial carcinoma (UC), pancreatic cancer, or cholangiocarcinoma. Results showed that some patients with HER2-positive UC or pancreatic cancer could benefit from T-DM1 treatment [40].
Inotuzumab ozogamicin (InO)The CD22 antigen, a 135-kDa type I transmembrane sialoglycoprotein, is found in the cytoplasm of nearly all B lineage cells and is specifically expressed on B cells. The CD22 antigen is predominantly expressed in IgM+ IgD+ B cells [41]. InO is an ADC drug created by conjugating the human IgG4 mAb targeting CD22 with the cytotoxic chemotherapeutic drug calicheamicin through an acid-unstable splice. The binding of InO to CD22-expressing tumor cells initiates endocytosis of the InO-CD22 complex, leading to hydrolysis of the N-acetyl-γ-khakimycin dimethylhydrazide junction. Activation of N-acetyl-γ-kadzimycin dimethylhydrazide induces double-stranded DNA breaks, subsequently causing cell cycle arrest and cell death [42]. InO plays a crucial role in the treatment of acute lymphoblastic leukemia (ALL) by targeting cancer cells that abnormally express CD22, thereby inducing cell cycle arrest and apoptosis [43].
InO seems to be an effective salvage measure for patients with advanced ALL, enabling more patients to undergo stem cell transplantation and achieve long-term survival [44]. A phase III trial of InO in relapsed/refractory ALL has been completed. In this study, patients treated with InO exhibited significantly higher CR rates, lower disease burden in remission, and longer duration of remission compared to the group treated with standard chemotherapy [45]. Meanwhile, patients treated with InO showed improved clinical outcomes and QoL [46]. A multicenter, parallel, open-label phase III trial was conducted to assess the efficacy of InO in adult patients with recurrent/refractory ALL. The results indicated a higher CR or CR with incomplete hematologic recovery rate in the InO group compared to the standard-of-care (SoC) group. The median OS was 7.7 months in the InO group and 6.2 months in the SoC group [47]. In a study evaluating the antitumor activity and safety of InO for the treatment of CD22-positive relapsed/refractory ALL, the results showed that all treated patients had a median PFS of 3.9 months and a median OS of 7.4 months. The most common AEs with any grade included neutropenia (28%), increased AST (26%), nausea (21%), vomiting (17%), fatigue (15%), and febrile neutropenia (15%) [48].
In a phase II trial, InO was investigated as a monotherapy in pediatric patients with relapsed/refractory ALL. The study included a total of 32 enrolled patients, with 28 receiving treatment, and 27 being evaluable for efficacy. The results revealed a 1-year EFS rate of 36.7% and an OS rate of 55.1% [49]. In a multicenter study focusing on low-dose post-transplant InO for preventing relapse in ALL, it was found that the maximum tolerated dose of InO was 0.6 mg/m2. The study reported a 1-year non-relapse mortality rate of 5.6%, a PFS of 89%, and an OS of 94% [50].
The detection of measurable residual disease stands out as a significant predictor of relapse in ALL. In a phase II study investigating InO for the palliation of measurable residual disease in ALL, the results indicated a 69% response rate, leading to measurable residual disease negativity. The 2-year relapse-free survival rate for the entire cohort was 54%, and the a 2-year OS rate was 60%. Most AEs were of lowgrade. Consequently, InO demonstrates favorable survival rates, measurable residual disease negativity, and safety for patients with ALL and measurable residual disease positivity [51].
Moxetumomab pasudotox (MP)Developed by AstraZeneca and granted FDA approval in 2018, MP is a recombinant immunotoxin comprising moxetumomab targeting CD22, a 38 kDa fragment of pseudomonas exotoxin A, and the linker mc–VC–PABC. It is utilized for treating adult patients with relapsed/refractory hairy cell leukemia (HCL) who have not responded to at least two systemic therapies (including purine nucleoside analogues). MP marks the first drug approved for HCL treatment in over 20 years [52].
The FDA approval of MP relies on data from the phase III clinical study, Study 1053, which was a single-arm, multicenter study involving 80 patients diagnosed with HCL or an HCL variant. These patients had undergone at least two systemic treatments. The treatment involved intravenous injection of 40 µg/kg MP on the 1st, 3rd, and 5th day of each 28-day cycle, totaling 6 cycles. The primary endpoint was CR, defined as achieving CR and maintaining hematologic remission for over 180 days. The data revealed that MP monotherapy achieved an ORR of 75%, a CR of 41%, and a durable CR of 30%. The most common AEs (grade 3–4) included decreased lymphocyte count (20%), asymptomatic hypophosphatemia (10%), and anemia (10%) [53]. Updated data confirmed that MP exhibited high durable response rates and a minimal residual disease negative rate in heavily pre-treated patients with HCL. It was deemed safe, manageable, and a new feasible treatment option [54].
Polatuzumab vedotin (PV)Developed by Genentech, PV is an ADC composed of the antibody CD79b linked to MMAE through a cleavable dipeptide linker (mc–VC–PABC). It received its initial approval for the treatment of adult patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who have undergone at least two prior therapies in conjunction with bendamustine and rituximab (BR) [55].
The approval was based on findings from an open-label, global, multicenter, phase Ib/II clinical study known as GO29365. In this study, 80 patients with relapsed/refractory DLBCL, who had previously undergone at least one treatment regimen, were randomly assigned to two groups. One group received BR with PV, while the other group received BR alone. Both groups underwent a total of six 21-day cycles of treatment. The study assessed CR rate as primary endpoint. Results demonstrated a higher CR rate in the BR with PV group compared to the BR alone group, with a significantly elevated CR rate evaluated by the independent review committee at the end of treatment (40.0% vs. 17.5%). In the BR with PV group, the most common grade 3–4 AEs included thrombocytopenia (41%), neutropenia (46.2%), infection and infestation (23.1%), and anemia (28.2%). Additionally, among transplant-ineligible patients with relapsed/refractory DLBCL, the BR with PV group exhibited a 58% lower risk of death compared to the BR group [56]. The phase III POLARIX study (NCT03274492) further demonstrated PV as an effective option for treating patients with relapsed/refractory DLBCL [57]. It revealed that PV in combination with rituximab plus cyclophosphamide, doxorubicin, and prednisone (Pola-R-CHP) significantly improved PFS compared to R-CHOP in both Asian and global populations, with comparable safety profiles between Pola-R-CHP and R-CHOP [58].
A preclinical investigation demonstrated that PV induces the degradation of the BCL-2 protein family member MCL-1 through the ubiquitin/proteasome system. When PV was used in combination with venetoclax and anti-CD20 antibodies obinutuzumab or rituximab, the targeted MCL-1 antagonistic effect led to tumor regression in preclinical non-Hodgkin lymphoma (NHL) models. Importantly, these regressions were sustained even after discontinuation of treatment. In the phase Ib clinical trial, severely pre-treated patients with recurrent or refractory NHL received the combination therapy of PV, venetoclax, and an anti-CD20 antibody. A significant proportion of patients responded positively to the treatment, with 76% of patients with follicular lymphoma and 29% of patients with DLBCL achieving either complete or partial responses [59].
In a phase Ib/II trial evaluating the safety and activity of mosunetuzumab plus PV in relapsed/refractory aggressive large B-cell lymphoma (LBCL), the best ORR was 59.2%, the CR rate was 45.9%, median PFS was 11.4 months, and median OS was 23.3 months. The most common grade ≥ 3 AEs were neutropenia and fatigue. These findings suggest that the combination of mosunetuzumab with PV exhibits good safety and a highly persistent response, making it suitable as a second-line treatment for patients with relapsed/refractory LBCL who are not eligible for transplant [60].
However, a single-arm, phase Ib/II study revealed that PV combined with rituximab and lenalidomide in treating patients with relapsed/refractory DLBCL did not meet the threshold of predetermined activity. The CR rate was 31%, and the most common grade 3–4 AEs were neutropenia and thrombocytopenia [61].
Enfortumab vedotin (EV)Nectin-4, a type I transmembrane protein, is notably overexpressed in various malignant tumors, including bladder cancer. Its overexpression plays a role in promoting tumor cell proliferation, differentiation, and invasion through the activation of the PI3K/AKT signaling pathway, contributing to malignant tumorigenesis, metastasis, and recurrence [62, 63]. Consequently, Nectin-4 has emerged as a promising target for systemic therapy in locally advanced or metastatic urothelial carcinoma (la/mUC). EV is an ADC that combines a human antibody against Nectin-4 with the cytotoxic MMAE through a cleavable junction. Upon binding to Nectin-4, EV forms a complex that internalizes within Nectin-4-expressing cells. The released MMAE binds to tubules, disrupting the cellular microtubule network and leading to cell cycle arrest and apoptosis [64].
In the phase I dose-escalation study EV-101, incremental administration of 1.25 mg/kg EV occurred on the 1st, 8th, and 15th day of a 28-day cycle. Results from the study involving 112 patients with mUC treated with single-agent EV showed an investigator-assessed confirmed ORR of 43%, with a DOR lasting 7.4 months. The median OS was 12.3 months, and the 1-year OS rate reached 51.8%. The most frequently reported treatment-related adverse events (TRAEs) with an incidence rate of ≥ 30% included fatigue, alopecia, decreased appetite, dysgeusia, nausea, peripheral sensory neuropathy, pruritus, and diarrhea [65].
In a two-cohort, single-arm, phase II study (EV-201) involving 125 patients with metastatic UC, a final ORR of 44%, a CR rate of 12%, and a median DOR of 7.6 months were confirmed. This demonstrated a more favorable treatment outcome compared to standard chemotherapy [66]. The EV-301 trial further showcased the ability of EV to prolong the OS of patients compared to standard chemotherapy, with a 30% reduction in the risk of death, as indicated by a hazard ratio (HR) of 0.70 (95% confidence interval[CI] 0.58–0.85). PFS also improved with EV compared to chemotherapy, with an HR of 0.63 (95% CI 0.53–0.76). The incidence of TRAEs was 93.9% for EV and 91.8% for chemotherapy, with the incidence rates of grade ≥ 3 AEs being 52.4% and 50.5%, respectively. AEs associated with EV were manageable [67].
Subsequent retrospective studies of EV monotherapy have demonstrated its effectiveness in treating individuals in important patient populations previously excluded from clinical trials, including those with conditions such as diabetes. This highlights the maturation of research on EV with a broader population of recipients [68].
The FDA granted breakthrough therapy designation to the combination of EV with pembrolizumab (EV + P), approving it as a first-line treatment for patients with la/mUC who are not suitable for cisplatin [64]. In a phase II trial study, cisplatin-ineligible patients received treatment with EV + P, leading to demonstrated tumor shrinkage in a majority of the patients [69]. EV + P showcased the preservation or improvement of QoL, functioning, and symptoms in cisplatin-ineligible patients with la/mUC. Notable and clinically meaningful improvements were observed in European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-Core Questionnaire (EORTC QLQ-C30) scores at weeks 12 and weeks 24 in the EV + P group. Additionally, there was a significant decrease in worst pain scores measured by the Brief Pain Inventory Short Form (BPI-SF). In patients receiving EV monotherapy, the overall QoL assessed by the EORTC QLQ-C30 remained stable. These findings suggest that both EV + P and EV monotherapy have a positive impact on QoL, functioning, and symptom management in patients with la/mUC who are ineligible for cisplatin-based therapy [70].
In another study involving patients with la/mUC ineligible for cisplatin therapy, EV + P demonstrated a high confirmed ORR and a persistent response as first-line therapy [71].
Trastuzumab deruxtecan (T-DXd)T-DXd is an ADC that combines trastuzumab (a humanized mAb targeting HER2) with an exatecan derivative (a topoisomerase I inhibitor) through a linker designed for the targeted delivery of cytotoxic agents into cancer cells. In comparison to T-DM1, T-DXd can deliver a higher payload of cytotoxic drugs, and its improved membrane permeability allows it to kill more tumor cells through the “bystander effect” [72, 73]. The FDA has approved T-DXd for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have previously received two or more anti-HER2 therapies [74].
In an open-label, dose-escalation, and dose-expansion phase I trial, investigators assessed the safety, tolerability, and activity of T-DXd in advanced solid tumors expressing HER2. The results revealed a confirmed objective response in 66 out of 111 patients, with disease control confirmed in 104 out of 111 patients. The median follow-up was 9.9 months, and the median time to response, DOR, and PFS were 1.6 months, 20.7 months, and 22.1 months, respectively. All patients experienced at least one TEAE. Common grade 3 or more severe TEAEs included anemia (17%), neutropenia (14%), leukopenia (9%), and thrombocytopenia (8%). Moreover, 19% of patients experienced at least one serious TEAE, and interstitial lung disease, organizing pneumonia, or pneumonitis occurred in 20 patients [75].
In the phase II DESTINY-Breast01 trial, T-DXd demonstrated sustained antitumor activity in patients with HER2-positive metastatic breast cancer who had previously received ≥ 2 anti-HER2 treatments, including T-DM1 [76]. Subgroup analysis from DESTINY-Breast01 indicated markedly improved patient outcomes when T-DXd was
留言 (0)