
記住我
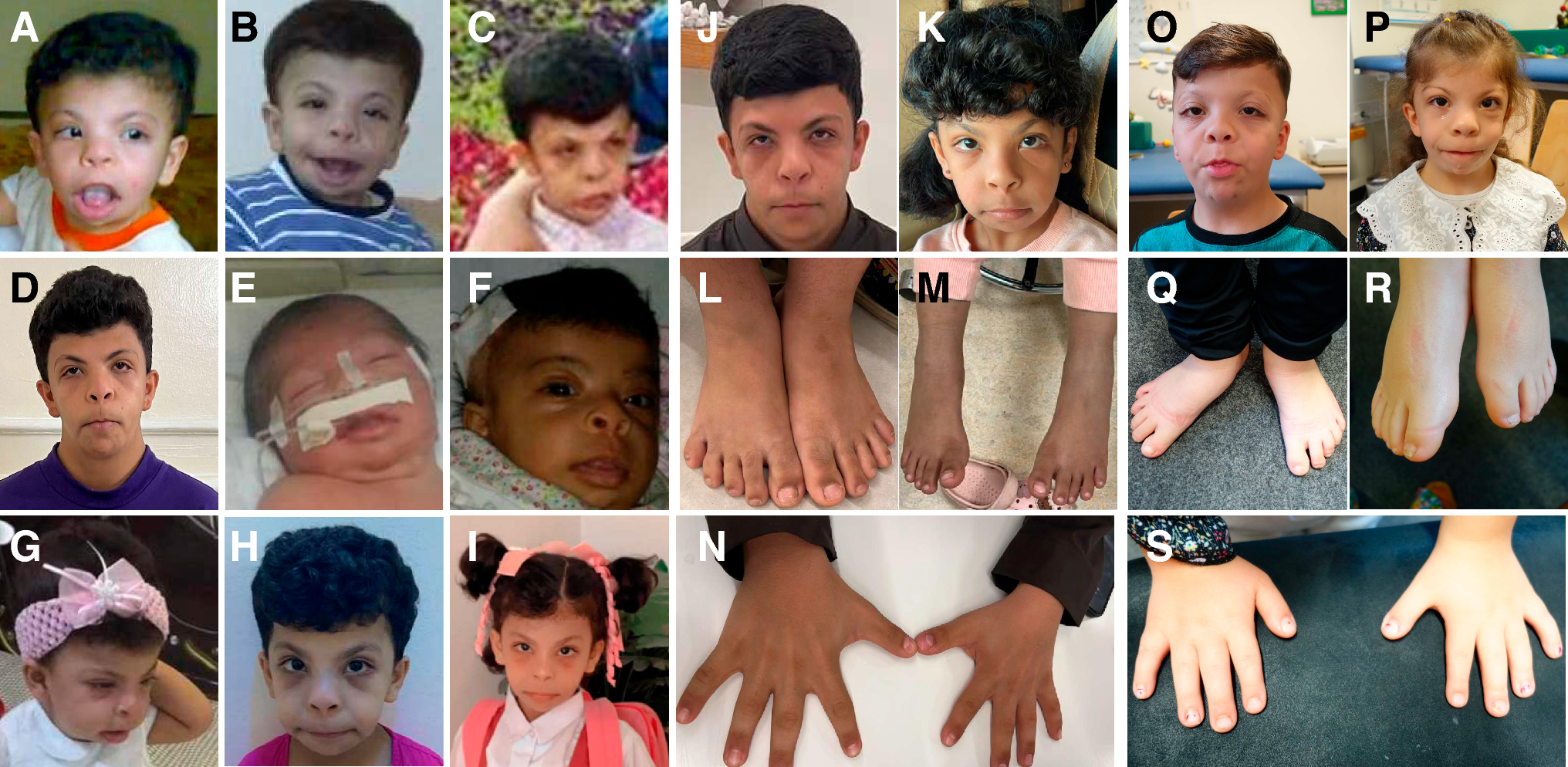
CAGI6 RGP organizers shared with all participants 35 solved cases for training (6 cases proband-only, the remaining trios, duos, and quartets). For each subject, the VCF file was provided. HPO terms and the known causative variant for each proband were also shared. A detailed description of proband’s phenotypes, causative variants and family composition is reported in Challenge datasets in Stenton et al. (Stenton et al. 2023).
As a preprocessing step, we filtered out variants with high allele frequency in gnomAD (common variants in populations with allele frequency greater than 0.05 according to gnomAD v3.0) and sequencing artifacts, i.e. variants detected in more than 13 alleles in the whole dataset provided within the challenge.
Ethical considerationsAll RGP participants engage in a consent video or video call with a trained research coordinator. During this interaction, participants review the study protocol, which includes provisions for sharing de-identified data, and they provide signed informed consent (Mass General Brigham IRB protocol 2016P001422). The RGP organizers executed an institutionally signed (Broad-Northeastern) data transfer agreement. As predictors in the CAGI 6 challenge, we were required to sign and adhere to a registered access model and the CAGI Data Use Agreement (genomeinterpretation.org/data-useagreement.html). All details are provided in Stenton et al. (Stenton et al. 2023).
Features engineeringWe developed our model to emulate the reasoning process of a geneticist overseeing the Variant Interpretation process, aimed at solving undiagnosed cases. During the machine learning model’s feature engineering phase, each feature was meticulously designed to ensure independence from both the disease and the associated phenotypic characteristics of the patient. To accurately mimic the real diagnostic workflow, we identified four essential levels of information necessary to determine whether a specific variant constitutes the molecular diagnosis for the patient.
The first level involves assessing the pathogenicity of the variant (as an example, according to the ACMG/AMP guidelines). This question can be addressed by determining the variant’s pathogenicity through a quantitative pathogenicity score.
The second level revolves around determining whether the variant explains the observed phenotypes in the patient. Quantitative measures of phenotypic similarity between the variant and the proband’s phenotypes are employed to answer this question.
The third level focuses on evaluating whether the variant segregates in the family based on a complete penetrance model. Additionally, it examines whether the observed segregation aligns with the inheritance pattern associated with the condition. In this case, it is essential to verify the actual or inferred segregation pattern and the expected inheritance patterns for the disease.
Finally, the variant must be reliable and not be a result of sequencing artifacts or a common variant in the studied population. Variant quality metrics can be leveraged to address this aspect.
This set of information will serve as features for our model.
By considering these four levels of information, our model emulates the reasoning process of a geneticist, aiding in the interpretation of variants and potentially leading to the identification of the molecular diagnosis for undiagnosed cases.
The list of features used in the model is reported in Table 1.
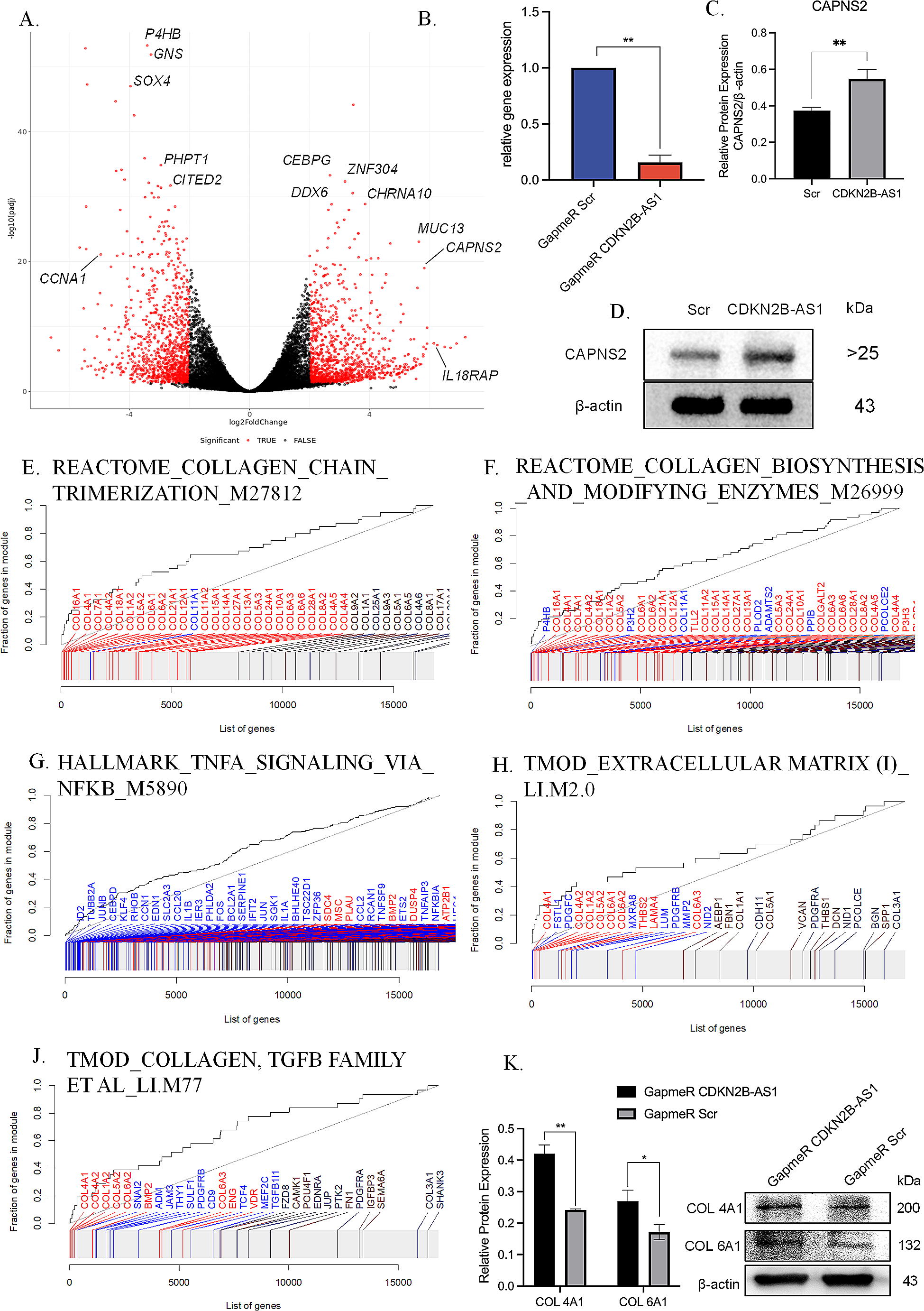
Table 1 Summary of the features exploited by the Suggested Diagnosis modelVariant pathogenicityTo assess variant pathogenicity, we analyzed the VCFs with eVai, the enGenome proprietary software that implements the ACMG/AMP guidelines. eVai assigns one of the five ACMG/AMP classes to each variant according to the implementation previously shared (Nicora et al. 2018). Additionally, eVai associates to each variant a pathogenicity score, which is predicted by a ML classifier based on the ACMG/AMP criteria triggered by each variant. We have previously developed a logistic regression model to predict variant pathogenicity based on ACMG/AMP standard guidelines (Nicora et al. 2022a), where the model was trained on bona-fide pathogenic and benign variants from the Clinvitae database. Hence, we assign the logistic regression predicted probability of pathogenicity (named pathogenicity score) to each variant. The ACMG/AMP classification is computed for all the variants on disease-associated genes, according to the principal databases, such as MedGen (https://www.ncbi.nlm.nih.gov/medgen/), Disease Ontology (https://disease-ontology.org/), and Orphanet (https://www.orpha.net/).
In our previous work, we showed that the eVai pathogenicity score had better performance in comparison with CADD, VVP, and a Bayesian approach on data from Clinvitae and the ICR639 project (Nicora et al. 2022a).
Phenotypic similarityFor each variant in a given gene, we calculated the Phenotypic Similarity between that gene and the proband’s phenotype. Computational analysis of phenotypes can greatly enhance the prioritization of variants. By computing measures of similarity between a patient’s clinical manifestations (provided as a set of HPO terms) and disease descriptions associated with genes, phenotype-based prioritization tools utilize standards and quantitative similarity measures to cluster and compare phenotype sets.
Phenotypic similarity scores are based on Human Phenotype Ontology information. Starting from standard phenotypic similarity indexes (e.g.: HPOSim package (Deng et al. 2015)) and exploiting the terms distance in the Direct Acyclic Graph (DAG) of the ontology, we developed a set of phenotypic similarity scores that take into account both the frequency of HPO terms in diseases and the specificity of the mutated genes in explaining sample’s phenotypes. More in detail, we firstly defined the HPO term sets associated with the gene and with the condition related to the gene as reported in Human Phenotype Ontology resources. We then exploited Resnik similarity between each HPO term of the patient and each term associated with the gene (or condition). Finally, we computed the phenotypic similarity between the two HPO sets, using the best-match average strategy (BMA), which calculates the average of all maximum similarities on each row and column of the similarity matrix S, as described in Eq. 1.
$$Sim_ \left( \right) = \frac^ max_ s_ + \mathop \sum \nolimits_^ max_ s_ }}$$
(1)
Equation 1: best-match average strategy to compute the similarity between 2 sets of HPO terms. g1 of size m includes HPO terms provided to describe the patient; g2 of size n includes HPO terms associated with the gene (or with the condition). sij is the Resnik similarity between i-th HPO term of g1 and j-th HPO term of g2.
The frequency of each HPO term in the condition (if reported in HPO resource) is used to weight each HPO term contribution in g2, when combining Resnik similarities in Eq. 1.
The final phenotypic score is a combination of the gene-based and condition-based phenotypic similarities computed as described above.
We performed a preliminary analysis on 35 training set samples to evaluate the phenotype-based prioritization capabilities of our phenotypic score against other common solutions. We evaluated the prioritization performance of various tools (Phen2Gene (Zhao et al. 2020), Amelie (Birgmeier et al. 2020), Phrank (Jagadeesh et al. 2019) and Phenolyzer (Yang et al. 2015)) by examining the ranking of the causative gene based on its phenotypic similarity with the clinical phenotypes presented by the patient. A list of all mutated genes and HPO terms was given as input to each of the tools. The computation of the phenotypic similarity score is limited to genes with known associations with phenotypes, according to the HPO database.
Inheritance informationTo encode inheritance information for each variant, we consider three key factors: the genotype of the variant, the expected inheritance mode of the disorder (e.g.: as reported in MedGen for the gene where the variant is located), and any available family segregation data (Licata 2023). Briefly, the expected mode of inheritance for each variant (such as autosomal dominant or recessive, X-linked, or de novo), is assigned based on the family segregation pattern (if available) and the variant’s genotype (Table 2). Secondly, we evaluate the expected inheritance mode of the condition associated with the gene where the variant is located. This evaluation is based on the inheritance mode reported in MedGen for the gene. If these two values match, this information is used to trigger a binary feature indicating whether the variant is inherited or not.
Table 2 Inheritance pattern description and assignment’s rules when family analysis is availableVariant qualityLastly, the quality of each variant is assessed and encoded. Higher-quality variants (as an example, in terms of filter, genotype quality, allelic balance, and coverage), are generally more reliable and carry stronger implications for disease association and inheritance.
According to this description, in the dataset built for the CAGI challenge, each instance represents a possible genetic diagnosis (a single variant or a combination of candidate compound heterozygous variants) and the features cover the four described levels of information. This feature had a dichotomous value (0/1), computed based on the value of the filter field reported for each variant. Specifically, a filter field value of “Pass” was indicated as Quality = 1, while any value other than “Pass” was indicated as Quality = 0 (Fig. 1).
Fig. 1
Workflow of the “Suggested Diagnosis” model
Training and evaluation strategyThe labeled dataset provided by the organizers consists of 35 solved cases (training set). Each training sample contains a median of 7700 variants, which were included in the training set, totaling approximately 269,000 variants overall. Among all these instances, the causative variants indicated by the data providers were labeled as pathogenic, while the remaining variants were labeled as benign. A detailed description of the 35 cases is reported in the CGI6 RGP Challenge publication (Stenton et al. 2023).
The decision to solely utilize the training set provided by the CAGI organizers as a source of information during the training phase is driven by the aim to maintain homogeneity between the training and test sets. These two provided datasets exhibit homogeneity in terms of experimental factors such as sequencing platforms and genomic targets. Additionally, they show similar overall variant numbers and distributions, including coding/non-coding variants proportion. The phenotypic descriptions are consistent and performed by the same pool of clinicians, eliminating any phenotype selection bias between the training and test sets. Furthermore, the distribution of features, such as pathogenicity scores and quality measures, is similar, as shown by the reliability analysis that we performed (described in the following section). However, to exclude the possibility that the model overfits the CAGI data, we assess the generalization capabilities of the model on an additional independent test set (the DDD dataset), described in the next paragraphs.
For the solved test cases, the true causative was not made available, to compare the performance of different teams in an unbiased way. Therefore, to perform model selection and to preliminary understand whether our approaches work well, we set up a Leave-One-Proband-Out Cross Validation (LOPO CV) on the training set. In detail, for each of the 35 training probands, we retained that proband for testing (LOPO test proband, containing a median of approximately 7700 variants), and we trained different models on the remaining 34 probands (training set with a median of approximately 261,800 variants). We evaluated the ranking performance on the LOPO test proband that was not included in the training. This procedure was repeated for each training probands, thus resulting in 35 trained models for each different machine learning classifier and 35 evaluated cases. Given the phenotypic heterogeneity characterizing the 35 probands, the LOPO strategy allows the assessment of the generalization capabilities of the machine learning models.
We compared the performance of different machine learning classifiers, all implemented in the scikit-learn package (Pedregosa et al. 2011), such as Bayesian classifier, linear models, ensemble models, and Multilayer perceptron. The best models were then trained on the complete training set and were used to predict the 30 test cases provided by the CAGI organizers.
Predictive uncertainty and reliability analysisIn addition to predicting the probability that each variant is disease-causing, CAGI organizers admit that also a standard deviation of the prediction can be submitted. Some classifiers, such as Bayesian classifiers, already embed a notion of uncertainty, while to calculate the uncertainty of the prediction for the ensemble models we exploited the framework suggested by Shaker et al. (Shaker and Hüllermeier 2020): we computed the uncertainty as Shannon entropy of the “weak” classifiers’ predicted probabilities, where the “weak” classifiers are the classifiers in the ensemble.
We’ve addressed this task by computing the uncertainty of predictions through the analysis of its reliability.
Machine learning generalization ability represents the ability of the classifier to maintain good performance not only on train/development data but also on different datasets that can be provided during deployment over time. Poor generalization ability of machine learning models in time and across different datasets has been widely reported, especially in healthcare, and it can hamper trust in machine learning prediction (Kelly et al. 2019). A possible cause for a decrease in performance is dataset shift when the variable distributions greatly differ from training. In the context of variant interpretation, it has been shown that the performance of in silico tools to predict variants’ damaging impact greatly varies and is affected by circularity and error propagation, when the same genes exploited to train the models are also used to evaluate them during tests (Grimm et al. 2015). Consequently, machine learning performance can decrease on new data. To understand if the prediction of a model trained on a particular set of data can be considered reliable, i.e. if we can trust such prediction, a reliability assessment can be performed. With the term “pointwise reliability” we refer to the degree of trust that a single prediction is correct. A simple criterion to determine whether a prediction may be reliable is the so-called “density principle”, that checks if the classified instance is similar to the training set. If so, we are more willing to trust the prediction in this instance, because the model may have successfully learned to classify data in the same features space and distribution of the training set (Nicora and Bellazzi 2020). This approach remains completely independent of the ground truth class of the tested instance, which is unknown in a real deployment case, as well as in the context of the CAGI 6 RGP challenge. Instead, it relies only on feature distributions. To ensure the trustworthiness of our models’ predictions, we implemented a reliability assessment framework capable of establishing if the test cases originate from a population similar to the training cases, and therefore if we could trust the predictions of our models. Specifically, we compare the feature distributions of the training set with those of the test set, employing a previously developed reliability assessment approach (Nicora and Bellazzi 2020; Nicora et al. 2022b). Briefly, for each feature, we calculated the “borders” in the training set (Olvera-López et al. 2010). Borders are defined as training instances in a class that exhibit the closest proximity to an example of the opposite class, for a given attribute, or that have the minimum/maximum value for that feature. For each variant in a test case, we compare its attributes with the corresponding training borders, and we record the number of attributes for which that variant would become a border if included in the training set. The higher this number, the more information this case would add to the training, and therefore the less it is similar to the current training. The reliability score is calculated as the following formula:
$$rel\left( x \right) = 1 - \frac }}$$
where \(_\) is the number of attributes for which the instance would become a border and \(m\) is the total number of attributes. Therefore, for each variant in each test case, we assign a reliability score from 0 to 1 according to the above formula. Scores closer to 1 indicate a higher degree of reliability. For each test proband, we calculated various statistics, such as the median and standard deviation of the reliability score, the minimum reliability score, and the percentage of variants that have a reliability score equal to 1.
留言 (0)