
記住我
The aim of the study is to investigate the effects of SNM on symptoms and quality of life in children and adolescents. We intend to demonstrate that SNM is safe and effective in treatment of constipation and FI refractory to conventional therapeutic options.
The following hypotheses will be investigated within 3 months of treatment:
(A)SNM is superior to ENM regarding treatment of abdominal pain as main symptom of refractory chronic constipation in childhood and adolescence.
(B)Further exploratory hypotheses will be evaluated:
SNM/ENM is effective and safe in reducing symptoms of chronic constipation and FI in children and adolescents as an additional therapeutic approach while continuing conventional treatment options unchanged.
SNM/ENM treatment is effective in the subgroup of patients with Hirschsprung’s disease (HD).
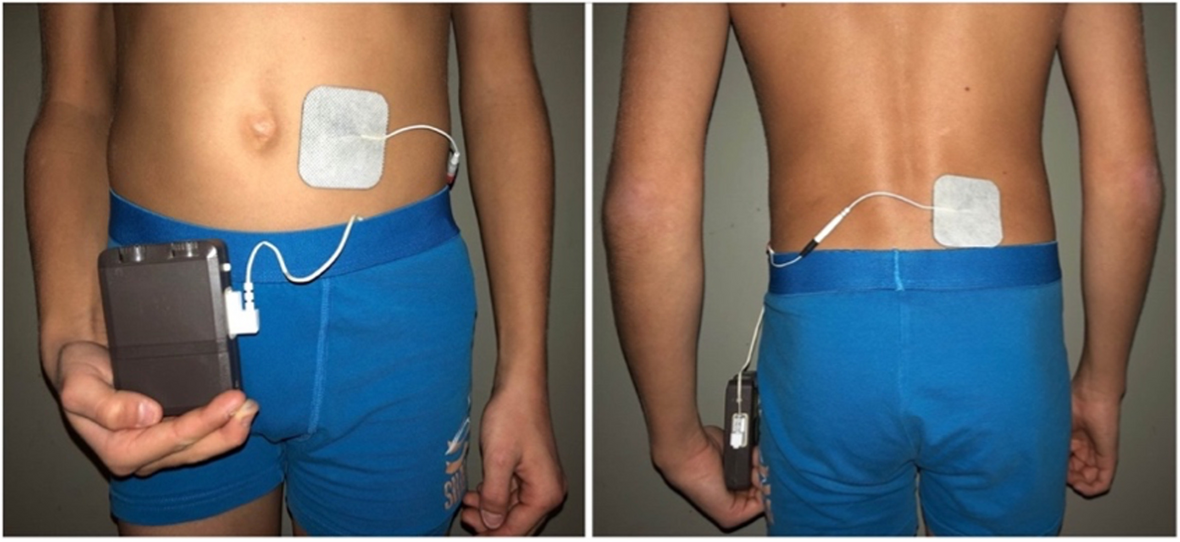
TechniqueEnteral neuromodulation (ENM)ENM is administered non-invasively via two cutaneous adhesive electrodes, placed paravertebrally and periumbilically (Fig. 1). The stimulation is applied with a frequency of 15 Hz and a pulse width of 210µs. Detailed stimulation techniques have been described previously [5] and are administered via a pulse generator (Ostimex® ProfiPlus TENS/EMS 335035) [11]. The adhesive electrodes (50 × 50mm in size) have a conductive surface and adhere to the skin by themselves. The manufacturer makes multiple use possible.
Fig. 1
Enteral neuromodulation. Placement of the cutaneous adhesive electrodes of this non-invasive approach is depicted: the electrical field is built between a paravertebrally and periumbilically placed electrode
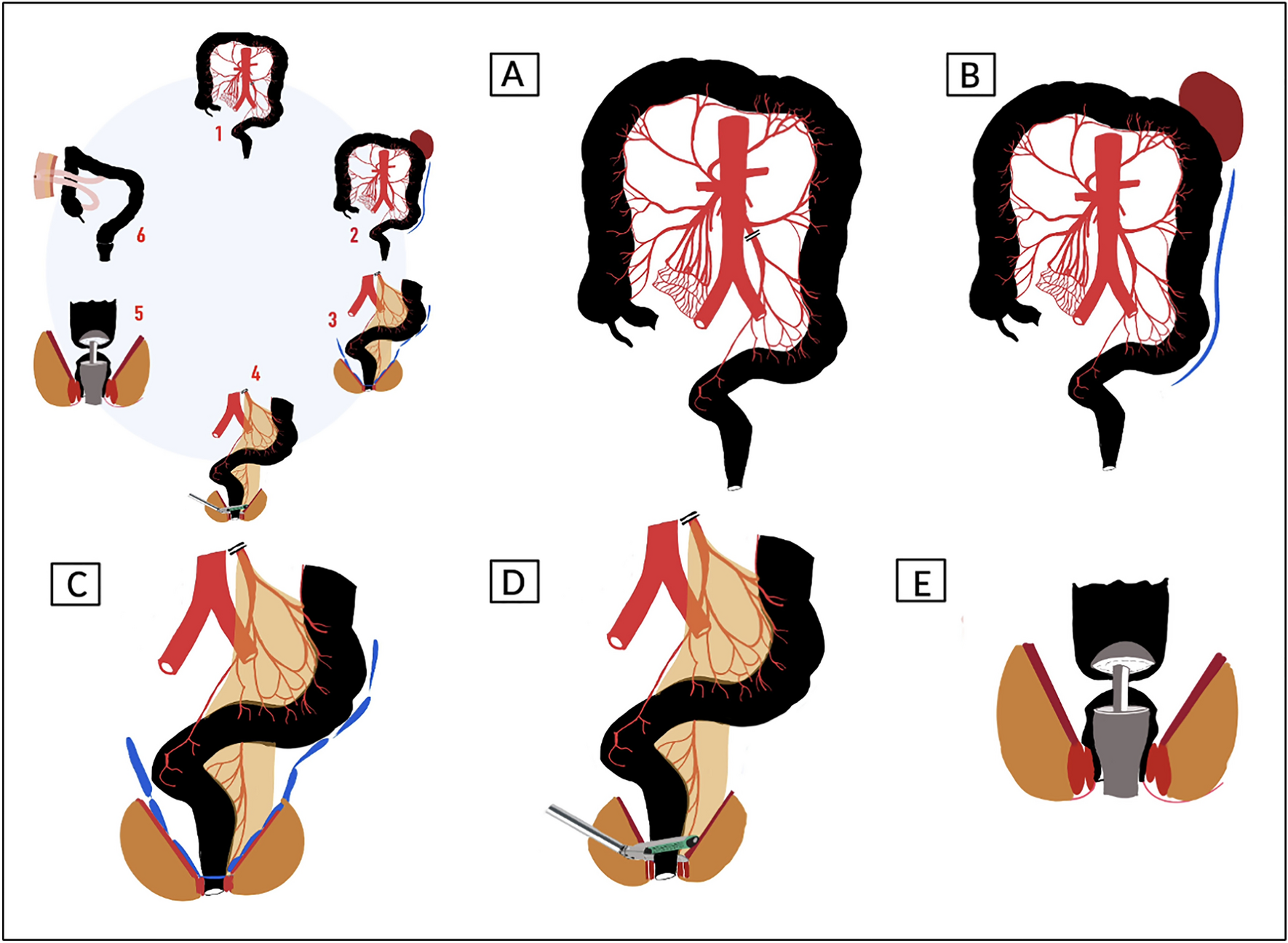
Sacral neuromodulation (SNM)SNM is applied via two surgical interventions. First, the flexible electrode lead with four equally spaced electrode contact points (quadripolar tined lead electrode, Medtronic Interstim, Fig. 2A) is inserted percutaneously via the sacral foramen close to the sacral spinal nerve S3 or S4. A motor or sensory response can be monitored directly intraoperatively by probatory electrical stimulation and determines the side of the final implantation of the tined electrode. Stimulation is generated externally by a pulse generator and will be started on the first postoperative day with a frequency of 14 Hz and a pulse width of 210µs. The bipolar electrical field is generated between different poles of the tined lead, directly located at the sacral spinal nerves.
Fig. 2
Sacral neuromodulation. Figure 2 illustrates the surgical implantation of sacral neuromodulation. A The tined lead electrode is placed at the sacral spinal nerve S3 or S4. B, C The previously implanted electrode is connected to a pulse generator, which is then implanted subcutaneously above the gluteal region
After a successful trial phase with a neuronal response, an implanted pulse generator (IPG, Medtronic® Interstim Micro) is connected to the implanted electrode (Fig. 2B) and subcutaneously implanted (Fig. 2C) 4 weeks after the electrode’s implantation to secure optimal electrode positioning and exclusion of postoperative complications.
Trial designThis is a monocentric, randomized, unblinded, 2-arm parallel-group trial in a 1:1 allocation ratio. The study is conducted at the University Hospital Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) from 2019 to 2023. Potential subjects are screened among patients attending pediatric surgical and pediatric gastroenterological clinics. Diagnoses are made according to the ROME IV criteria for FC [12]. Interventions are provided to subjects based on their group assignment of either 3 months of SNM or ENM therapy. Regular checkups and a follow up appointment are conducted within the trial and after 6 and 12 months after the initiation of the intervention. All participants are informed of the purpose of the study and obtain their informed consent before participating in this parallel group study. The subjects may choose to withdraw from the study, or they may be withdrawn from the study, at any time at the discretion of the investigator. If a subject withdraws or is withdrawn, every effort is made to complete and report the observations. The study design is presented in Fig. 3.
Fig. 3
Study design. Figure 3 illustrates the design of the study, highlighting the comparison of the two study groups at different time points
Study population: eligibility criteriaInclusion criteriaTo be eligible for participation, subjects are required to fulfill the following criteria:
age between 2–17 years
informed consent
chronic constipation according to the ROME IV criteria for more than 3 months with abdominal pain and with or without FI [12] despite underlying diseases such as FC, rectal evacuation disorders or HD.
refractory to conventional treatment in an appropriate weight-adapted application (training for bowel movements, lifestyle changes, pelvic floor training, pharmacological options)
in cases of HD: diagnosis confirmed histologically by rectal biopsies and in case of resection of an aganglionic segment: period between surgery and SNM at least 1 year
in cases of anorectal malformation or mechanical obstruction: post-surgical status: period between surgery and SNM at least 1 year
Exclusion criteriaExclusion criteria are determined as follows:
metabolic, inflammatory, and hormonal causes for chronic constipation
toxic megacolon or further emergencies, which must be treated surgically
sacral fractures or malformations prohibiting SNM access to target nerves
inflammatory bowel diseases
external rectal prolapse
neuronal malignancies under medical and radiation therapy
seizures
RecruitmentParticipants are recruited from the outpatients and inpatients clinics of the pediatric surgery and gastroenterology departments. Two or more experts determine the eligibility according to the inclusion and exclusion criteria. Informed consent, especially on treatment modalities and off-label use of SNM/ENM in childhood and adolescence, must be obtained before enrollment in the trial by next-of-kin and participants (6 years and older). Thereafter, a baseline assessment is performed, collecting all baseline information relevant to the study (sociodemographic data, medical history, current defecation status). The study schedule is summarized in Fig. 4 and included as the SPIRIT template as Fig. 5. At all timepoints, patients are able to withdraw their participation and end therapy with neuromodulation.
Fig. 4
Study schedule. Figure 4 explains evaluation of outcome variables at different time points of the study
Fig. 5
SPIRIT figure. Figure 5 summarizes the schedule of enrolment, interventions, and assessments according to the SPIRIT figure
RandomizationParticipants are enrolled in the study groups at a 1:1 allocation to either the SNM or ENM group. Furthermore, patients are allocated 1:1 stratified based on HD/FC. Enrollment of subjects (assessment of eligibility, patient contact to obtain informed consent) is done by investigators and clinicians prior to randomization. The study design does not allow blinding of the therapist or further analyses in clinical follow-ups. The design is open label with only outcome assessors being blinded, so unblinding will not occur. Data analysis and statistical assessment are conducted separately and anonymously. The statistician performing the statistical analyses is blinded to individual group allocation and treatment.
InterventionsNon-pharmacological treatment in both study groupsAll subjects are provided with non-pharmacological management counseling prior to the study inclusion, including lifestyle modifications, such as (1) age-appropriate fluid consumption, (2) balanced diet and (3) regular sportive activity (at least 3 × 30min per week). Education and conduction of toilet training is further advised to improve regular bowel movements. If patients show further symptoms, irresponsive to these measurements, enrollment is conducted and non-pharmacological treatment is continued throughout the trial.
Pharmacological treatment in both study groupsPharmacological options are included in the study population as follows: disimpaction with polyethylene glycol (PEG, 1.5-2mg/kg/d for 1–2 weeks) and initiation of maintenance therapy (PEG, 0.2–0.4 mg/kg/d) are conducted prior to the study inclusion. Maintenance therapy is continued throughout the study period in both groups. Supportive local applications such as saline enemas or stimulant laxatives (glycerin, bisacodyl) are applied as needed. Change of pharmacological treatment is not recommended during the study period, especially in case of rectal enemas. Medication due to other diagnoses is not changed during the study period.
Group 1: enteral neuromodulationENM is continuously applied for 12 weeks in each patient. Stimulation intensity is individually set by each patient to achieve a comfortable stimulation below pain threshold.
Group 2: sacral neuromodulationWith the implantation of the electrode, stimulation of S3/4 is continuously applied. Patients are able to set stimulation intensity parameters within a preset range via an external device with or without the IPG.
Statistical methodsObjectivation and data validationSpecialized questionnaires were developed to objectivize symptoms and medical history of patients. These include sociodemographic data, medical history, and current defecation status and were adjusted for time points of “Baseline”, “Treatment with ENM” and “Treatment with SNM”.
Quality of life data are assessed according to the ‘Revised Children’s Quality of Life Questionnaire’ (KINDLR) before and after treatment. This reliable and validated questionnaire is a self-report measurement for health-related quality of life in children and adolescents [13, 14]. It consists of 24 5-point Likert-scale items, covering 6 quality of life dimensions: physical well-being, emotional well-being, self-esteem, family, friends, and daily functioning (school or nursery school/kindergarten). Items are partially reversely scored and linearly transformed to a 0 to 100 scale according to the manual. The sub-scales of these six dimensions are combined to produce a total score. Higher scores indicate a better quality of life. The questionnaire is available in three age-specific versions (The Kiddy-KINDLR for 4–7 years of age, the Kid-KINDLR for 8–12 years of age and the Kiddo-KINDLR for 13–16 years of age).
Data collectionData is extracted from the bowel movement diaries and specialized questionnaires. Recorded data is stored in case report forms at a secured place. Data is coded and entered in electronical files using Excel 2007 software (Microsoft Cooperation) by at least 2 different data administrators to reduce mistakes. All files are protected with password, which is only known by the investigators. Only the investigators have access to the final trial dataset. The information of the grouping and the results of the study are provided to the participants after the trial. Publications will only report aggregated data, and personal identities will not be disclosed.
Sample size calculationAs there is still insufficient clinical evidence on neuromodulation treatment in children and adolescents with FC and HD, this study requires only a small sample size. Power analysis is based on abdominal pain as primary outcome variable. A minimum of 78 subjects in total and 39 subjects per group is powered to detect a difference of at least 30%, with a 95% confidence interval (α = 0.05) and statistical power of 80% (β = 0.2).
Statistical analysisIBM SPSS version 28 (IBM, Armonk, NY) is used to perform statistical analyses by an independent and blinded statistician. Continuous variables are presented as mean ± standard deviation, categorical data are reported as frequencies and percentages. If data losses occur during the study, the last observation is carried forward to adjust the missing data in follow-up evaluations. If large amounts of data are missing in one patient, the patient will be excluded from the study based on the decision of the project management group. If participant’s withdrawal is observed in a high number of patients, protocol modifications will be made in regular meetings. We compare clinical outcome data using chi-square and Fisher’s exact tests at defined time points pre- and post-treatment in both groups. Quality of life data is compared using unpaired or paired, two-tailed sample t-tests, if applicable. In case of multiple analyses, adjustment of p-values will be conducted accordingly. We set the confidence interval to 95% and all p-values less than 0.05 are considered indicative of statistical significance.
Ethical approval and registrationThe study protocol was approved by the local ethics committee (Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), No. B18_20) and complied with the Declaration of Helsinki. Written informed consent is obtained from each subject and next-of-kin by the investigators before the subject enters the trial. The study is registered with clinicaltrials.gov (Identifier NCT04713085). If protocol modifications are required, amendments will be submitted to and approved by the local ethics committee. To disseminate our findings, the clinical trial results will be published in peer-reviewed journals. Additionally, the study’s protocol is in congruence with the SPIRIT 2013 statement [15]; the SPIRIT checklist has been added to this publication.
Organizational aspects of the trialThe coordinating project management group evaluates the trial’s progress and publishes necessary reports. Communication between ethics committee, patient groups and members of the trial’s staff is provided by the principal investigator, being the head of the coordinating project management group (meetings 4 per year). The trial steering committee coordinates organizational matters and documentational aspects (meetings 1 per month). Performing medical investigators meet once a month and are providing day by day support and patient’s contact. If protocol modifications are required, sponsors and funders are notified first by the project management group. The participating center will be informed based on a revised protocol, which will be sent as soon as the principal investigator, sponsors and funders are in agreement. Changed amendments will furthermore be submitted to and approved by the local ethics committee and updates will be uploaded at clinicaltrials.gov. Any deviations from the protocol will be fully documented using a breach report form. There are no external auditing trials planned, as this is a low-risk intervention.
In case of technical problems of applied devices with consecutive recalls, the trial is terminated by the principal investigator and interim results might then not be published.
Patient and public involvementThe study supports patient and public involvement to improve the study’s design and outcome variables. Based on suggestions from patients and patients’ organizations at the beginning of the trial, inclusion criteria, documentation of data and outcome variables were modified. Further suggestions are repeatedly evaluated in regular meetings of the study’s head and possibly implemented as modification (see above).
Study outcomesParticipants are required to complete bowel movement diaries throughout the trial to the first target point. Children aged up to 7 years are thereby represented by their next-of-kin, whereas participants ≥ 13 years of age are advised to fill in the questionnaires by themselves. Between age 8 to 12, documentation is conducted together and depending on the child’s autonomy. At routine check-ups at baseline, 4 weeks, 3 and 6 and 12 months, specialized questionnaires must be completed. Quality of life analysis is conducted at baseline and after 3, 6 and 12 months, as described below (Fig. 4).
Primary outcomes and measurementPrimary outcome variables are defined, and change is measured between time points of baseline, 3 and 6 months of therapy as follows:
(1)Episodes of abdominal pain: Number of episodes of abdominal pain per week
(2)Episodes of FI: Number of episodes of FI per week
(3)Defecation frequency: Number of bowel movements per week
(4)Stool consistency: Daily assessment of Bristol Stool Scale [16]
Patients are classified based on treatment response and efficacy. A “clinically relevant” improvement is defined in cases with at least 2/4 fulfilled criteria, in which symptom control or -reduction is achieved.
Secondary outcomes and measurementSecondary outcome variables are defined as follows:
(1)Improvement of proprioception: as mentioned in the specialized questionnaires
(2)Episodes of urinary incontinence: the number of episodes per week is evaluated with the criterion for a clinically relevant improvement in cases of reduction by at least 50% of episodes per week.
(3)Quality of life: assessment based on the KINDLR questionnaires
(4)Safety of treatment: adverse events as mentioned in the specialized questionnaires
FI is diagnosed at the age of ≥ 4 years in cases of prior adequate toilet training and urinary incontinence at the age of ≥ 6 years with at least 4 episodes per week.
Safety analysis and adverse eventsFormer studies on neuromodulation confirm a good safety profile. The participants conduct self-administered neuromodulation therapy at home. The participants are informed about potential adverse events and if any occur, they are instructed to terminate therapy and to immediately communicate with the researchers. Safety problems are reported to the clinical authorities (head of department, ethics committee) as well as to the manufacturers themselves. Appropriate treatment changes are then initiated. Adverse events are additionally recorded as part of the data collection for each session and will be reported to the clinical authorities and the manufacturers.
留言 (0)