Cells and viruses
HEK-293, 293 T, Huh7, MDCK, and VeroE6 cells were all cultured in growth media (Dulbecco’s modified Eagle medium, 10% fetal bovine serum, and antibiotics) and incubated at 37 °C and 5% CO2. SARS-CoV-2 wild-type SH01 strain (GenBank: MT121215), Delta, BA.2, BA.5.1, and BA.5.2 were obtained from the Chinese Center for Disease Control and Prevention after isolation from patients in Shanghai, China. The pH1N1 influenza virus was provided by the Shanghai Public Health Clinical Center of Fudan University (Shanghai, China). The pH1N1 virus was created using reverse genetics and contained six gene segments from the A/PR8 virus and HA and NA genes from A/Califomia/7/2009.
Mouse
Six- to eight-week-old female C57BL/6 J and K18-hACE2 transgenic mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and GemPharmatech Co., Ltd. (Jiangsu, China), respectively. All experiments involving live SARS-CoV-2 were conducted in the animal biosafety level 3 (BSL-3) facilities of Fudan University following International Standard Operating Procedures. All influenza virus experiments were performed in the BSL-2 facilities of the Shanghai Public Health Clinical Center of Fudan University following the standard operating protocols approved by the Institutional Biosafety Committee at the Shanghai Public Health Clinical Center, Fudan University. All animal studies that did not involve pathogenic pathogens were performed in SPF facilities, with the approval of the Institutional Animal Care and Use Committee of Tianjin Medical University (Tianjin, China).
Vaccine construction
The recombinant AdC68 vector was engineered by deleting the E1/E3 gene and replacing the E4-orf6/7-orf4 gene with AdHu5 equivalents. The Beta-Alpha chimeric SARS-CoV-2 RBD dimer consisted of one Beta RBD (spike protein residues 319–541, GenBank: MZ410518.1) and one Alpha RBD (S protein residues 319–541, GenBank: OM443028.1) connected in tandem repeats. Omicron-Delta chimeric SARS-CoV-2 RBD dimer comprises one Omicron RBD (spike protein residues 319–541, GenBank: OP315382.1) and one Delta RBD (S protein residues 319–541, GenBank: ON908696.1) connected in tandem repeats. The tandem conserved TCEs from ORF1, ORF3, and M proteins of the ancestral SARS-CoV-2 are shown in Supplementary Table 1. The full-length HA gene sequence was based on pH1N1 (GenBank: ACP41953.1). These codon-optimized genes encoding the aforementioned transgene were inserted into the ∆E1 region of the AdC68 vector under the transcriptional control of the human cytomegalovirus promoter and the SV-40 polyadenylation sequence.
AdC68-HATRBD was packaged, rescued, and amplified in HEK293 cells and subsequently purified by cesium chloride density gradient ultracentrifugation. The infectious units of recombinant adenoviruses were titrated on HEK293 cells using QuickTiter™ Adenovirus Titer Immunoassay Kit (Cell Biolabs, San Diego, CA, USA) following the manufacturer’s instructions.
Immunization and challenge
Mice were anesthetized with intraperitoneal tribromoethanol and then intranasally vaccinated with 5 × 107 IFU of AdC68-HATRBD in a 15 μl volume. In the heterologous vaccination experiments, 5 μg of ZF2001 or 3 μg of QIV through i.m. route was administered. The control group received a placebo (PBS). At the end point of experiments, mice were euthanized by cervical dislocation and the BALF, spleens, and/or lungs were harvested. In Figs. 2, 3, and 6, each dot represents data from two mice. That is to say, the specimens were mixed from two mice, including BALF, serum and lymphocytes. In other animal experiments, each point represents data from one mouse.
Challenge was carried out with 1 × 105 FFU of BA.5.2 or 10 × LD50 of pH1N1 administered intranasally. Mice were monitored daily for clinical signs and weight loss, with 70% of their initial weight considered a humane endpoint. To measure viral loads, the trachea and half a fraction of lung tissue were harvested on day 5 post-infection and immediately frozen in dry ice and stored at −80 °C until processed. The other half of the lung tissue was fixed in 4% paraformaldehyde for hematoxylin-eosin staining.
Western blot
HEK293 cells were transduced with 105, 106, or 107 IFU of AdC68-HATRBD. AdC68-empty-transduced (107 IFU) HEK293 cells were used as controls. Twenty-four hours post-infection, the transduced cells were harvested and boiled at 100 °C with 1 × reductive loading buffer for 15 min. Western blotting was performed using anti-SARS-CoV-2 RBD antibody (1:2000 dilution, 40592-T62; Sino Biological, Beijing, China) or anti-influenza A HA antibody (1:5000 dilution, 86001-RM01; Sino Biological, Beijing, China), followed by HRP-conjugated anti-rabbit IgG antibody (1:5000 dilution, ab6721; Abcam, Cambridge, UK). β-actin was selected as the internal control (1:5000 dilution, HRP-60008; Proteintech, Wuhan, China). Blots and gels derived from the same or side-by-side experiments were processed together. The un-cropped and unprocessed western blot images are provided as Supplementary Fig. 4.
Enzyme linked immunosorbent assay (ELISA) for binding antibody measurements
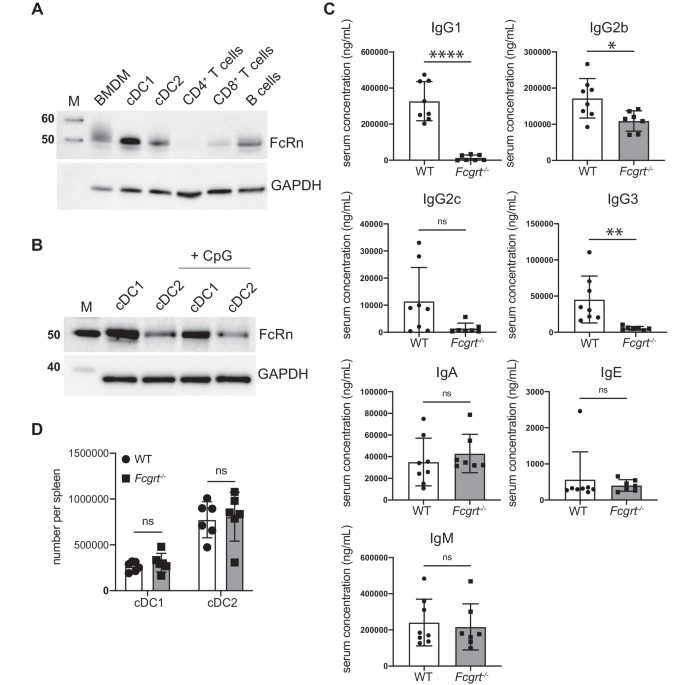
The IgG and IgA antibody levels in mouse serum and BALF were measured by ELISA, as previously described55. Briefly, ELISA plates were coated overnight at 4 °C with RBD protein of WT or BA.5 strain or HA protein of pH1N1 (100 ng/well, Sino Biological, Beijing, China). For lgG detection, serum samples were threefold serially diluted with 1:400 starting dilutions, and BALF samples were twofold serially diluted with 1:5 starting dilutions. For IgA detection, BALF samples were twofold serially diluted using an undiluted stock solution as the starting dilutions. Following 2-h incubation at 37 °C, secondary antibodies were added: HRP-conjugated anti-mouse IgG antibody (1:100000 dilution, ab6789; Abcam, Cambridge, UK); HRP-conjugated anti-mouse IgA (1:5000 dilution, 1040-05; Southern Biotech, Birmingham, AL, USA). Plates were again incubated for 1 h at 37 °C, followed by the addition of TMB substrate (NCM Biotech, Suzhou, China). Sulfuric acid (2 M H2SO4) solution was used to stop the reaction. The optical density (OD)450–630 was recorded using a microplate reader (Tecan, Männedorf, Switzerland). The binding antibody endpoint titer was determined as the reciprocal of the highest serum dilution that yielded an absorbance greater or equal to 0.1 OD unit above the absorbance of the pre-immune samples.
Production of SARS-CoV-2 pseudoviruses
SARS-CoV-2 pseudoviruses were produced as previously described78,79. Briefly, the backbone plasmid pNL4-3.Luc.R-E and pCAGGS-S-CΔ19 expressing spike of the WT strain or its variants were cotransfected into HEK 293 T cells by polyethyleneimine (Polysciences, Warrington, PA, USA). The spike amino acid sequences of WT strain and its variants were based on GISAID EPI_ISL_402124 (Wuhan/WIV04/2019), EPI_ISL_712096 (Beta variant), EPI_ISL_2029113 (Delta variant), EPI_ISL_6640916 (BA.1 variant), EPI_ISL_8515362 (BA.2 variant), EPI_ISL_13241867 (BA.5 variant), EPI_ISL_15812431 (BQ.1 variant), or EPI_ISL_14917761 (XBB.1 variant). The supernatant was collected 48 h post-transfection, filtered through 0.45 µm filters, and concentrated overnight with PEG 8000 at 4 °C.
Pseudovirus titration was performed based on HIV-1 p24 antigen quantification utilizing Lentivirus Quantitation Kit (Beijing Biodragon Immunotechnologies Co., Ltd, Beijing, China) following the manufacturer’s instructions; 1 × 108 lentivirus particles per well were used for the pseudovirus neutralization test.
SARS-CoV-2 pseudovirus neutralization assay
Huh7 cells were seeded at a density of 2.5 × 104 cells per well in opaque 96-well flat-bottom plates. Twenty-four hours later, each serum sample was threefold serially diluted and mixed with an equal volume of pseudoviruses. The mixture was incubated at 37 °C for 1 h before adding to Huh7 cells. Relative luciferase activity (RLA) was measured after 48 h using the SteadyGlo Luciferase Assay System (Promega, Madison, WI, USA). The pVNT50 titer was calculated as the reciprocal of the highest serum dilution at which RLA was reduced by 50% compared to the RLA in virus control wells infected with pseudovirus in the absence of mouse serum. The lower limit of detection (LLD) of the neutralization assay was 20, and measurements below the LLD were assigned half the LLD.
SARS-CoV-2 neutralization assay
Serum samples were threefold serially diluted in 96 well U-bottom plates. An equal volume of SARS-CoV-2 consisting of WT strain, Delta, BA.2, BA.5.1, or BA.5.2 was then added to the diluted serum. The serum-virus mixture was incubated at 37 °C for 1 h before transferring to 96-well plates with 4 × 104 VeroE6 cells per well. Plates were incubated at 37 °C, 5% CO2 for 48 h, and fixed with 4% paraformaldehyde in PBS for 20 min. The plates were washed and incubated with rabbit anti-N antibody (1:2000 dilution, A18797; Abclonal, Wuhan, China), followed by incubation with peroxidase-conjugated anti-rabbit IgG (1:1000 dilution, AS014; Abclonal, Wuhan, China) and peroxidase substrate (Seracare, Milford, MA, USA). Virus-infected cell foci were counted using an ImmunoSpot microanalyzer (CTL, Shaker Heights, OH, USA). The FRNT50 titer was measured as the reciprocal of the highest serum dilution at which foci were reduced by 50% relative to the control wells infected with SARS-CoV-2 in the absence of mouse serum. The LLD of the neutralization assay was 10, and measurements below the LLD were assigned half the LLD.
Influenza virus neutralization assay
The influenza virus-specific neutralization activity of mouse serum was analyzed using a micro-neutralization assay in MDCK cells. A two-fold serial dilution of serum samples was performed in a 96-well plate and incubated with 100 tissue culture infectious dose 50 (TCID50) of pH1N1 at 37 °C for 1 h. The MDCK culture media was then replaced with the mixture and incubated at 37 °C for 48 h. The culture supernatants were then cultured with same volume of 1% chicken red blood cells diluted in PBS and incubated for 20 min at room temperature. The neutralizing titer was the highest dilution of serum that inhibited virus-induced hemagglutination. The LLD of the neutralization assay was 10, and measurements below the LLD were assigned half the LLD.
Haemagglutination-inhibition assay
Serum samples were treated with a receptor-destroying enzyme from Vibrio cholerae (Denka Seiken, Tokyo, Japan) at 37 °C overnight, followed by heat-inactivation at 56 °C for 30 min. Serial serum dilutions were mixed with four hemagglutination units of inactivated pH1N1 virus in 96 well V-bottom plates for 1 h. The 1% chicken red blood cells diluted in PBS were added to each well, and the wells were incubated for 15 min before analysis. The reciprocal of the highest serum dilution that prevented complete hemagglutination was considered the HAI titer. The LLD of the HAI assay was 10, and measurements below the LLD were assigned half the LLD.
Adenovirus neutralization assay
As previously described, 5 × 104 cells per well A549 were seeded in 96-well overnight. A threefold dilution series of the test sera were mixed with an equivalent volume of 1 × 107 vp AdC68-Luc. After 2 h of incubation at 37 °C, the mixture was added to A549 cells. Two days later, RLA was measured. The NT50 titer was calculated as the reciprocal of the highest serum dilution at which RLA was reduced by 50% compared to the RLA in virus control wells infected with AdC68-Luc in the absence of mouse serum. The LLD of the neutralization assay was 10, and measurements below the LLD were assigned half the LLD.
Flow cytometry
As previously described40,80, the mononuclear cells from lungs and spleens were isolated and stimulated with 2 µg/mL of the RBD, TCEs, or HA pool in the presence of GolgiPlug (BD Biosciences, San Jose, CA, USA) for 12 h. The RBD peptide pool contained 15-mers overlapping by 11 amino acids derived from the BA.5 variant. The TCEs peptide pool contained 24 epitope peptides (Supplementary Table 1). The HA peptide pool contained 15-mers overlapping by 11 amino acids derived from pH1N1. Upon stimulation, cells were blocked with Fc block (1:250 dilution, 14-0161-85; BD Biosciences, Franklin Lakes, NJ, USA) and simultaneously stained with LIVE/DEAD Fixable dye (Thermo Fisher Scientific, Waltham, MA, USA) at 4 °C for 30 min. Cells were then stained with four surface markers containing PerCP/Cyanine5.5-CD3ε (1:500 dilution, 100328; BioLegend, San Diego, CA, USA), APC-Cy7-CD4 (1:250 dilution, A15384; Thermo Fisher Scientific, Waltham, MA, USA), Alexa Fluor 700-CD8a (1:250 dilution, 100730; BioLegend, San Diego, CA, USA), and FITC-CD44 (1:250 dilution, 48-0621-80; Thermo Fisher Scientific, Waltham, MA, USA) for 30 min at 4 °C, followed by fixation and permeabilization using fixation/permeabilization solution (BD Biosciences, San Jose, CA, United States) following the manufacturer’s instruction. Fixed/permeabilized cells were incubated with a cocktail of fluorescently labeled cytokine antibodies containing APC-IFN-γ (1:250 dilution, 505810; BioLegend, San Diego, CA, USA), Brilliant Violet 421-TNF-α (1:500 dilution, 506328; BioLegend, San Diego, CA, USA), PE-IL-13 (1:250 dilution, 159403; BioLegend, San Diego, CA, USA), and PE-Cy7-IL-4 (1:250 dilution, 504118; BioLegend, San Diego, CA, USA) for 30 min at 4 °C. Finally, samples were fixed with 2% PFA for 30 min before the acquisition, then acquired on a BD LSRFortessa (BD Bioscience, San Jose, CA, USA) and analyzed using FlowJo v10.
ELISpot assay
Single cells of spleens and lungs were isolated to determine the number of cytokines-producing cells using Mouse IFN-γ ELISpotBASIC kit and Mouse IL-4 ELISpotBASIC kit (MabTech, Nacka Strand, Sweden) according to the manufacturer’s instruction. Briefly, Multiscreen IP ELISpot plates (Millipore, Billerica, MA, USA) were coated with respective capture antibody overnight at 4 °C. After blocking and washing, 1 × 106 cells and 2 µg/ml HA pool were added to each well of the plate, and incubated for 48 h at 37 °C with 5% CO2. Spot counts were performed using an ImmunoSpot microanalyzer (CTL, Shaker Heights, OH, USA). The results are presented as the number of spot-forming unit (SFU) per well.
Viral load determination
The loads of SARS-CoV-2 and influenza virus in the tracheas and lungs were quantified using quantitative real-time-polymerase chain reaction from the RNA samples prepared as described above. For SARS-CoV-2, two sets of primers were used to detect the N gene of the viral genome and the E gene of sgRNA.
N Forward: 5′-GACCCCAAAATCAGCGAAAT-3′;
N Reverse: 5′-CTGGTTACTGCCAGTTGAATCTG-3;
N-probe: FAM-ACCCCGCATTACGTTTGGTGGACC-TAMRA;
E Forward: 5′-CGATCTCTTGTAGATCTGTTCTC-3′;
E Reverse: 5′-ATATTGCAGCAGTACGCACACA-3′;
E-probe: FAM-ACACTAGCCATCCTTACTGCGCTTCG-TAMRA.
SARS-CoV-2 viral loads were expressed on a log10 scale as viral copies per gram after calculation with a standard curve.
For influenza virus, the matrix (M) gene amplicon was quantified using the following primers:
M Forward: 5′-AAGACCAATCCTGTCACCTCTGA-3′;
M Reverse: 5′-CAAAGCGTCTACGCTGCAGTCC-3.
Ct values were normalized to the calibrator mouse gene GAPDH.
Genome was extracted from lungs and nasal turbinates using TIANamp Genomic DNA kit (Tiangen, Beijing, China). The genome load of AdC68-HATRBD was quantified by RT-qPCR using the Roche LightCycler® 480 system. The primer sequences to detect the hexon gene of AdC68 were:
Hexon Forward: 5′-GCCCTTCCACATCCAGGTGC-3′;
Hexon Reverse: 5′-CCATGGGGAAGAAGGTGGCG-3′;
AdC68-HATRBD load was expressed as viral copies per gram after calculation with a standard curve.
Histopathology
PFA-fixed lung tissues were embedded in paraffin, sectioned into 4 µm-thick, and stained with hematoxylin and eosin. According to previous study41, histopathological changes were scored from 1 to 5 based on the degree of interalveolar edema, intra-alveolar hemorrhage, and neutrophil infiltration.
Statistical analysis
All data are presented as means ± SEM unless otherwise stated, and asterisks and pound signs in the figures indicate statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). All experimental data used to compare differences among groups were analyzed via one-way ANOVA with Tukey’s multiple comparisons test using GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.

留言 (0)