Materials
SiO2-NPs ranging from 10 to 20 nm in size, dimethyl sulfoxide (DMSO) (#M81802), and 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl-2H-tetrazolium bromide (MTT; # M2128) were obtained from Sigma–Aldrich (St. Louis, MO, USA). [3H]-D-aspartate was purchased from PerkinElmer (Boston, MA). Cell culture medium was from Thermo Fisher Scientific (Carlsbad, CA), and plasticware was purchased from Corning (New York, NY).
Cell Culture and Silica Nanoparticles Stimulation Protocol
Endothelial cells form the primary structure of the BBB since these cells control the passage of molecules inside and outside the brain (Alahmari 2021). The barrier function of the endothelial cells is mainly provided by tight junctions (TJs) and several transport systems (Sweeney et al. 2018). As it was mentioned before, the BBB is a complex set of cells in which astrocytes participate actively, for example, astrocytes clear neurotransmitters (Danbolt 2001), produce glutathione (Dringen et al. 2015), synthesize and release trophic factors (Nuriya and Hirase 2016), and contribute to neurovascular coupling by extending end-feet processes to the vasculature (Abbott et al. 2006). Results from co-culturing experiments demonstrate that brain endothelial cell contact with astrocytes is required for GLT-1 and GLAST transporter expression (Lee et al. 2017).
Thus, several human cell lines have been used in BBB studies (Eigenmann et al. 2013; Weksler et al. 2013), but the most characterized are human cerebral microvascular endothelial cells (hCMEC/D3) (Weksler et al. 2013) and human brain microvascular endothelial cells (hBMEC) (Eigenmann et al. 2016). However, these models grow with a cocktail of adjuvants, and it is known that adjuvants have an impact on transporter expression (Eisenblätter and Galla 2002; Wedel-Parlow et al. 2009). So, in this work, we chose a human brain endothelial cell line (HBEC-5i) that can grow in a monolayer and mimic the BBB (Puech et al. 2018). Furthermore, this cell line has been cultivated with human astrocytes due to an improvement of the barrier properties (Abbott et al. 2006) by the close interactions between brain endothelial cells and astrocytes (Abbott et al. 2006; Helms et al. 2016); thus, these cell lines could represent the BBB and how the SiO2-NPs might disrupt it.
HBEC-5i endothelial cells and U-87MG astrocyte cells were obtained from ATCC; no. CRL-3245 and HTB-14, respectively, Manassas, VA, USA. Initially, HBEC-5i were cultured in Dulbecco’s Modified Eagle Medium, (DMEM-F12 HAM, no. 12400–016, Gibco), supplemented with 10% fetal bovine serum (FBS), 40 µg/ml microvascular growth supplement (MVGS; no. S00525, Gibco), and 1% of antibiotic solution, and U-87MG cells were cultured in Dulbecco’s Modified Eagle Medium, (DMEM-F12 HAM), supplemented with 10% fetal bovine serum FBS and 1% of antibiotic solution. Both cell lines were cultured, seeded, and stimulated separately. The HBEC-5i cell line was cultured on dishes coated with 0.1% gelatin (no. G2500, Sigma–Aldrich), incubated at 37 °C for ≥ 40 min, and then gelatin was aspirated before adding cells to the dishes. For the transport assays, to avoid the cells being detached, the HBEC-5i cell line was also seeded on dishes coated with 0.1% gelatin. Confluent monolayers of both cells (HBEC-5i and U-87MG) were treated with SiO2-NPs diluted in DMEM-F12 containing 0.5% FBS, at different concentrations and periods detailed below, based on the data of invitro experiments about SiO2-NPs neurotoxic effect (Orlando et al. 2017; Wang et al. 2011). The dilutions of SiO2-NPs were previously sonicated before treating the cells, by using a bath sonicator at room temperature for 15 min at 40 W to avoid SiO2-NPs agglomeration, as it was described previously (Rodríguez-Campuzano et al. 2020).
Methods
Cell viability was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (MTT; # M2128), which determines the ability of metabolically active cells to produce a purple formazan salt after the cleavage of the tetrazolium ring of a yellow substrate (MTT) by mitochondrial reduction (Denizot and Lang 1986). The amount of formazan was determined at λ = 560 nm and it is directly proportional to the number of viable cells. Briefly, HBEC-5i and U-87MG cells were seeded in 96-well plates (1 × 105 cells/well) and cultured to an 80 to 90% confluence; cells were treated with vehicle (control), different SiO2-NP concentrations (0.4, 4.8, 10, and 20 µg/ml), and periods (3 and 6 h) at 37 °C. Then, 3 h before the SiO2-NPs treatment ended, 20 µL/well of an MTT stock solution (0.5 mg/ml) was added directly into each well, and the plates were returned to the incubator. Finally, the medium was discarded, and 180 µL of DMSO was added to each well to dissolve the formazan crystals. Absorbance was measured with a microplate reader (Epoch, BioTek Instruments, VT, USA). Cell viability was calculated as follows: cell viability (%) = average OD of treated wells/average OD of control wells. Three independent experiments (n = 3) were performed in quadruplicate from three different passages.
Neutral Red Uptake Assay
This assay was performed as described previously (Repetto et al. 2008). This test is based on the use of a cationic probe (neutral red) which is taken up into cells by membrane diffusion where it becomes an ion trapped within the lysosomal compartment. Briefly, both cell lines were plated in a 96-well culture plates (1 × 105 cells/well) and treated with vehicle (control); different SiO2-NP concentrations ranging from 0.4, 4.8, 10, and 20 µg/ml, for 3 and 6 h; then, the medium of stimulation was discarded; and the cells were washed with 150 µl PBS per well. One hundred microliter of the neutral red medium was added to each well. The plates were incubated for 2 h at the appropriate culture conditions (37 °C). After that, the neutral red medium was removed; the cells were washed with 150 µl PBS, per well; and the washing solution was removed by gently tapping. Neutral red destain solution (50% ethanol 96%, 49% deionized water, 1% glacial acetic acid) was added (150 µl) per well, and the plate was shaken rapidly on a microtiter plate shaker for 10 min until obtaining a homogenous solution. The absorbance of dye was measured using a microplate reader at a wavelength of 570 nm. Three independent experiments (n = 3) were performed in quadruplicate from three different passages.
[3H]-D-Aspartate Uptake
The uptake of [3H]-D-aspartate (used as a non-metabolizable analogue of L-glutamate) was performed as previously described (Ruiz and Ortega 1995). Cells were seeded in 24 (5 × 105 cells/well) or 48 well plates (2.5 × 105 cells/well). Briefly, the medium was replaced with a pre-warmed uptake buffer containing 25 mM HEPES, 130 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 33.3 mM glucose and 1 mM NaH2PO4, pH 7.4, and 0.4 μCi/mL [3H]-D-aspartate ([3H]-D-Asp) (specific activity: 12.2 Ci/mmol, Perkin Elmer, MA, USA) (50 μM final aspartate concentration). Uptake was finished by the addition of ice-cold uptake buffer, and cells were lysed with 0.1 N NaOH for 2 h at room temperature. Aliquots of 10 μL were used for protein quantification, and the samples were transferred to scintillation vials, a liquid scintillation cocktail, and 50 μL of glacial acetic acid (to quench chemiluminescence) was added, and radioactivity was measured in a scintillation counter (PerkinElmer, MA, USA). Radioactivity counts were adjusted for protein quantity and calculated as [3H]-D-aspartate pmol/(mg protein min−1). Three independent experiments (n = 3) were performed in quadruplicates (4 wells by condition or group) from three different passages.
Glutamate Transport System Characterization
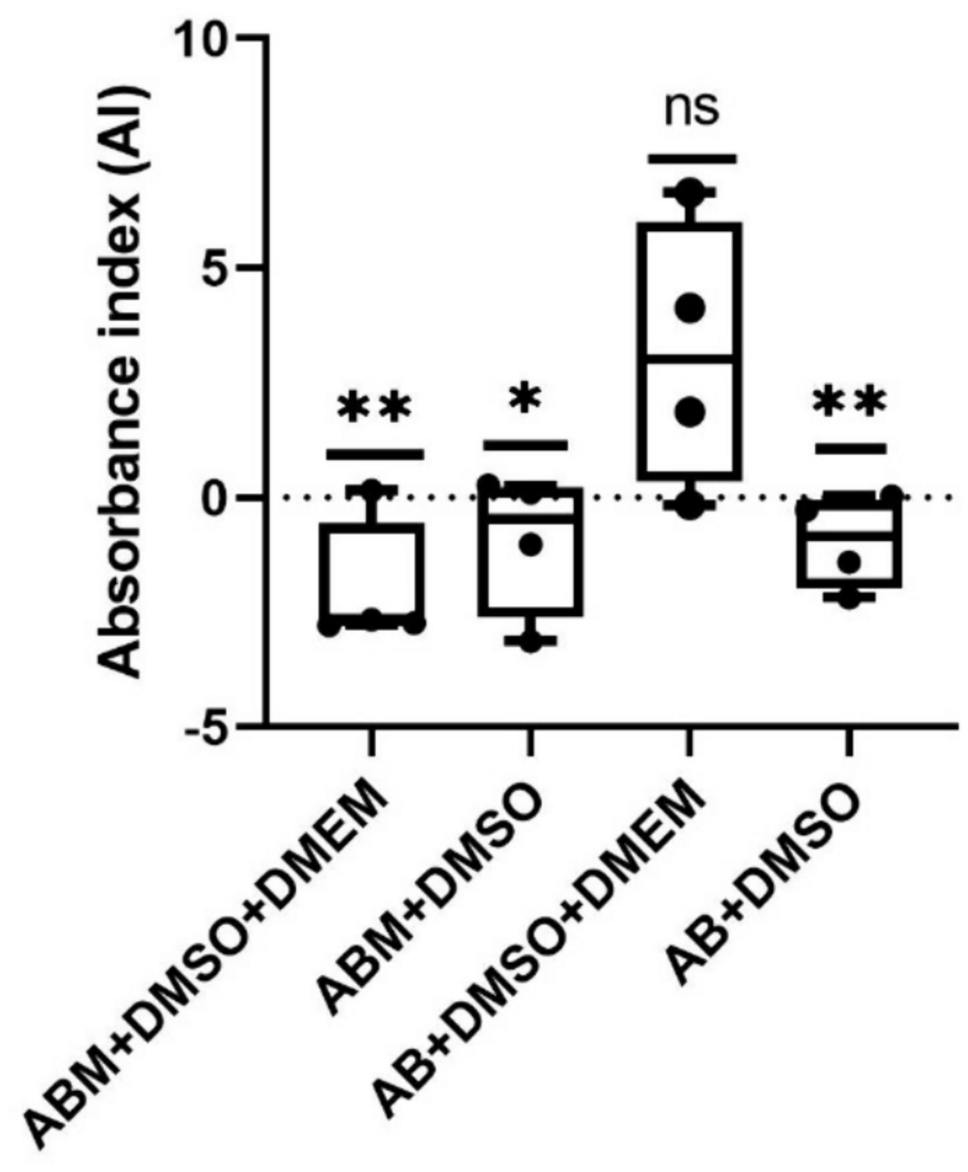
In the case of pharmacological characterization of the glutamate transport system, the cells were pre-treated for 30 min with selective excitatory amino acid transporter (EAAT) 2 blocker, dihydro kainic acid, (DHK) 100 μM, and TBOA 100 μM, a non-specific potent inhibitor of EAAT1,2, and 3. Also, we used glutamate (10 μM, 100 μM, 500 μM, and 1 mM) or aspartate (Asp 1 mM), because it has been shown their substrates downregulate the activity of excitatory amino acid transporters. Then we measured the uptake of [3H]-D-aspartate as we indicated previously, in the presence or absence of sodium (Na+/Na−, Figs. 2 and 3) since glutamate transport is electrogenic (Grewer and Rauen 2005). Three independent experiments (n = 3) were performed in quadruplicates from three different passages.
Kinetic Parameters of the Glutamate Transport System
For the determination of the kinetic constants, Km and Vmax, both cell lines were treated with uptake buffer containing 0.4 μCi/mL [3H]-D-aspartate + different unlabeled D-Asp concentrations 0,10, 25, 50, 100, and 200 μM (Sigma–Aldrich, MO, USA) (Fig. 4a and b) or pre-treated with DHK 100 μM 30 min before replacing the medium with [3H]-D-aspartate + different unlabeled D-Asp concentrations (Fig. 4c and d). Uptake was stopped after 30 min of incubation by washing the cells with an ice-cold uptake buffer, and the samples were processed as described above. A robust nonlinear regression was used to fit a model to the experimental data and estimated the parameters of the Michaelis–Menten equation (GraphPad Prism Software, La Jolla California, USA). Three independent experiments (n = 3) were performed in quadruplicates (4 wells by condition or group) from three different passages (Figs. 4 and 5).
Effect of SiO2-NPs on Glutamate Transporter Systems
To evaluate the effect of SiO2-NPs on glutamate transporter systems, we used different concentrations of nanoparticles (2.4, 4.8, 6.4, and 10 μg/ml) which have been demonstrated to be the closest physiologically relevant to SNC exposure (Xie et al. 2010; Wu et al. 2011). Also, in our group, Rodríguez–Campuzano et al. showed that exposure to SiO2-NPs at these doses affects protein synthesis in glial cells (Rodríguez-Campuzano et al. 2020). Recent studies have reported that exposure to SiO2-NPs activates a pro-inflammatory response, oxidative stress, and unfolded protein production (Wang et al. 2011; Wu et al. 2011; Nemmar et al. 2016), which results in cell death in the CNS, leading to an increase in the release of glutamate, over-activating its receptors, and saturating the excitatory amino acid transport system, triggering an ion imbalance that proceeds neuronal lysis, lasting in cell death cascades (Davide et al. 2018). Indeed, in a pilot experiment, we observed a decrease in the [3H]-D-aspartate uptake after the exposure of SiO2-NPs 4.8 μg/mL (data not shown). So, both cells were treated with a vehicle (control); Asp 1 mM; or different concentrations of SiO2-NPs (2.4, 4.8, 6.4, and 10 μg/ml) for 30 min. After the treatment, cultures were incubated with uptake buffer containing 0.4 μCi/mL [3H]-D-aspartate + unlabeled D-Asp 50 μM (Sigma–Aldrich, MO, USA). Uptake was stopped after 30 min of incubation by washing the cells with an ice-cold uptake buffer, and the samples were processed as described above. Four independent experiments (n = 4) were performed in quadruplicates from four different passages.
Some experiments were performed in the presence or absence of DHK (100 μM), and TBOA (100 μM) was pre-incubated 30 min before being exposed to SiO2-NPs (4.8 μg/mL), vehicle, or Asp 1 mM. After the treatment, the cultures were incubated with uptake buffer containing 0.4 μCi/mL [3H]-D-aspartate + unlabeled D-Asp 50 μM (Sigma–Aldrich, MO, USA). Uptake was stopped after 30 min of incubation by washing the cells with an ice-cold uptake buffer, and the incorporated radioactivity was evaluated as was mentioned previously. Four independent experiments (n = 4) were performed in quadruplicates from four different passages.
Effect of SiO2-NPs on Kinetic Parameters
The kinetics parameters were evaluated after treating the cells with 4.8 μg/mL of SiO2-NPs for 30 min. Then, the medium was replaced with uptake buffer containing 0.4 μCi/mL [3H]-D-aspartate + different unlabeled D-Asp concentrations 0,10, 25, 50, 100, and 200 μM (Sigma–Aldrich, MO, USA). Finally, uptake was stopped after 30 min of incubation by washing the cells with an ice-cold uptake buffer, and the samples were processed as described above.
A robust nonlinear regression was used to fit a model to the experimental data and estimate the parameters of the Michaelis–Menten equation (GraphPad Prism Software, La Jolla California, USA). Three independent experiments (n = 3) were performed in quadruplicates (4 wells by condition) from three different passages.
Statistical Analysis
Results are expressed as the mean ± SEM from a least three independent cultures. A one-way or two-way analysis of variance was carried out to determine significant differences between conditions followed by Dunnett’s multiple comparison or Bonferroni test, according to the results. For statistical analysis of kinetic experiments, t-tests were used. The probability of 0.05 or less was considered statistically significant. All the plots and analyses were performed with GraphPad Prism Software (La Jolla California, USA).




留言 (0)