
記住我
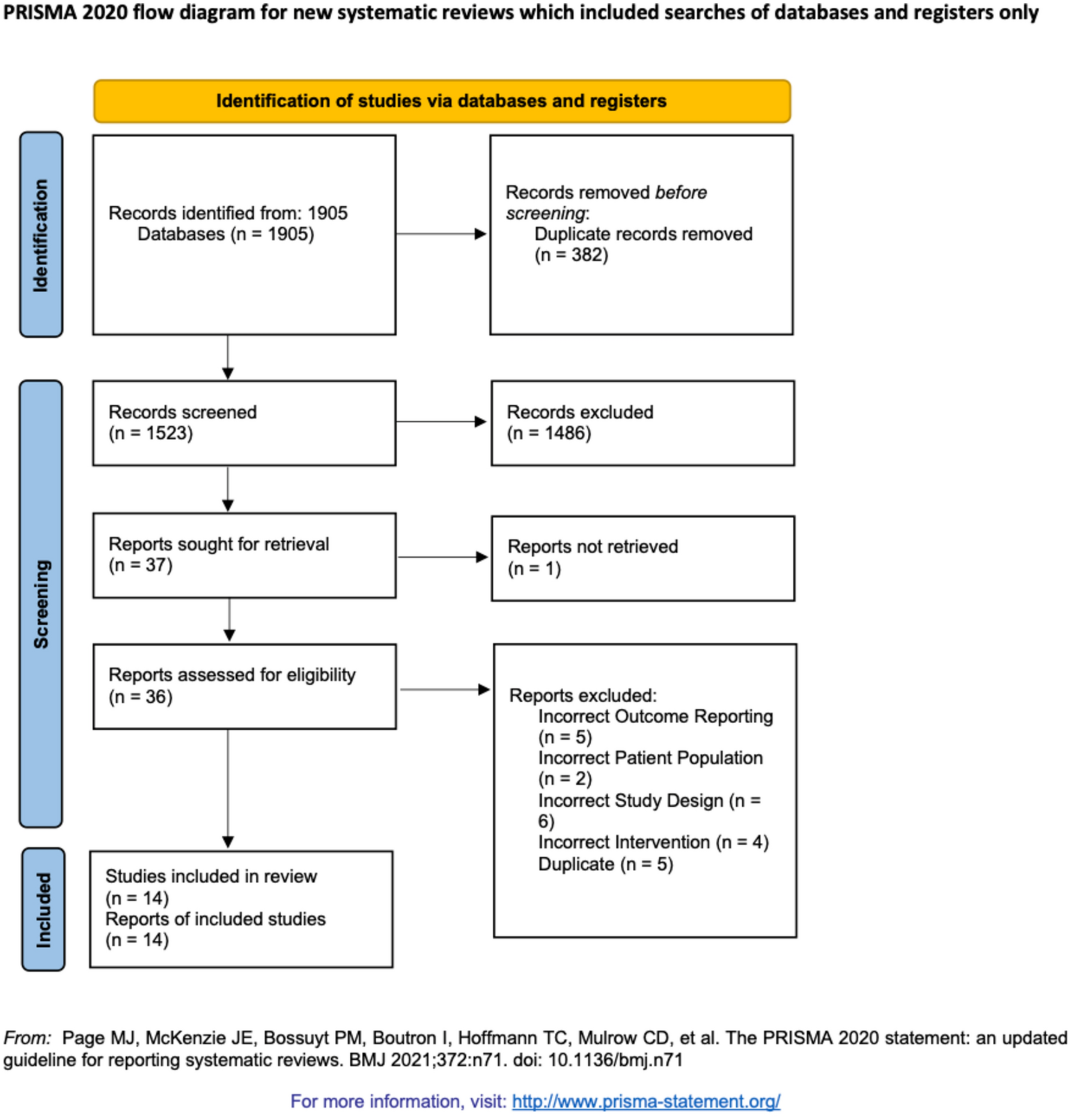
This study protocol is written in accordance with the SPIRIT guidelines [21, 22]. The SPIRIT checklist is provided in a supplementary file 1.
ObjectivesThe primary objective of these two multicentre international randomized studies is to determine which anastomosis, handsewn (end-to-end versus Kono-S anastomosis) or side-to-side stapled anastomosis after ileocolic resection for Crohn’s disease is superior with respect to endoscopic recurrence. Secondary objectives include evaluating gastrointestinal function, postoperative morbidity, quality of life and costs.
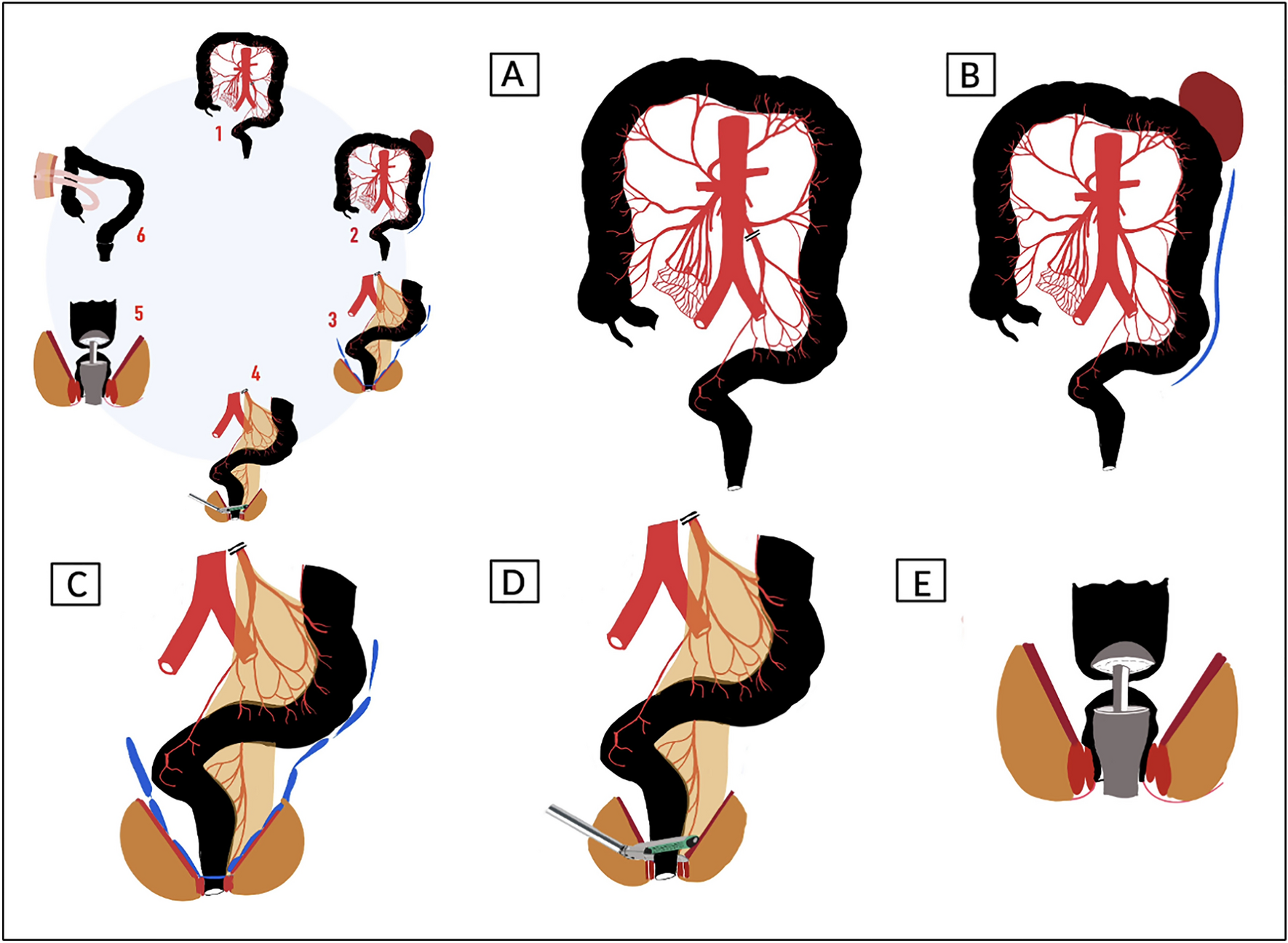
Study designThe HAND2END and End2End studies are two separate studies with similar designs conducted respectively in both Italy and the Netherlands. Three Italian centres and 12 centres in the Netherlands will participate in this prospective study. Both studies are multicentre randomized controlled superiority trials consisting of two phases. In phase one the two handsewn anastomoses (Kono-S and end-to-end) will be compared with the side-to-side stapled anastomosis. In this phase, participants will be randomized in a 2:1 ratio between (1) handsewn end-to-end (Fig. 1) or Kono-S anastomosis (Fig. 2) and (2) stapled side-to-side anastomosis (Fig. 3). If the interim analysis shows superiority of the handsewn anastomosis, phase two of the trial will compare the end-to-end (Fig. 1) with the Kono-S anastomosis (Fig. 2). In the Netherlands, three academic and eight non-academic teaching hospitals have agreed to participate in this national initiative. This multicentre (international) collaborative will greatly facilitate the (inter)national implementation of the study results. The attached video clips will demonstrate one way of performing these anastomoses (Supplementary files 2, 3, 4).
Fig. 1
Handsewn end-to-end anastomosis
Fig. 2
Handsewn Kono-S anastomosis
Fig. 3
Stapled side to side anastomosis
Study populationAll patients diagnosed with CD undergoing primary ileocolic resection or resection of recurrent disease of the neoterminal ileum will be considered for inclusion. To be eligible, a subject must meet all of the following criteria: adults of either sex, age ≥ 16 years or ≥ 18 years (in the Netherlands or Italy, respectively), diagnosed with either disease of the (neo)terminal ileum or ileocolic disease (L1 and L3 disease), previously confirmed by endoscopy with an indication for resection. Patients need to have a recent update of imaging (for example, ultrasound, MR/CT enterography) before surgery, the clinical case should have been discussed at the local multidisciplinary meeting and patients need to be able to comply with the study protocol and provide written informed consent. A potential subject who meets any of the following criteria will be excluded from participation in this study: inability to provide informed consent, patient under the age of 16 or 18 years (in the Netherlands or Italy, respectively), clinically significant medical condition within 6 months before the operation (for example, myocardial infarction, active angina, congestive heart failure, or other condition that would, in the opinion of the investigators, compromise patient’s safety), history of cancer of less than 5 years that might influence the patient’s prognosis, emergent operation, pregnancy or breastfeeding.
Ethical considerationsThe trials will be conducted according to Good Clinical Practice guidelines and the principles of the Declaration of Helsinki (2013). The End2End study was approved by the Medical Ethical Committee of the Amsterdam UMC, Amsterdam (MEC 2022.0533). The protocol is registered by the Dutch Central Committee on Research Involving Human Subjects (NL81981.018.22). The HAND2END study was approved by the Medical Ethical Committee of the IRCCS San Raffaele, Milan (EC-13/06/2022, version 2.0).
Informed consent procedureAll eligible patients referred for outpatient consultation for surgical resection will be screened for inclusion criteria during outpatient clinic visits. Patients who meet all the inclusion criteria will be informed on the details of the study and the possibility of participating in the trial. The informed consent procedure includes explanation of the study, the provision of a patient information sheet and ample time for addressing questions before signing the study consent form prior to the surgical procedure. Written informed consent is obtained by a participating surgeon, resident or trained research nurse before any study procedures. All included patients will be registered in Castor EDC and will be assigned a six-digit study number. A log of the assigned subject number will be maintained by each study site.
RandomizationRandomization of consented study participants to either handsewn (end-to-end or Kono-S) or stapled side-to-side anastomosis will be done using an online-based system for allocation concealment (Castor EDC). Allocation concealment will be ensured, as the service will not release the randomization code until the patient has signed for informed consent and has been recruited into the trial. Randomization will be stratified for primary or repeated ileocolic resection. The randomization block sizes will not be disclosed, to ensure concealment. There is no blinding of treatment allocation for the treating surgeon. The treatment will be blinded for all other physicians (for example, gastroenterologists, central readers) and patients. A statistician blinded to treatment allocation will analyse the data.
Study outlinePreoperativeAdult patients with ileocolic Crohn’s disease (CD) or recurrent disease of the terminal ileum will be screened during outpatient clinic visits. Qualified subjects expressing interest will be offered participation in the trial following multidisciplinary team discussion and will sign a written consent form if they agree to be enrolled. Medical treatment will be advised to be discontinued before surgery: glucocorticoids will be weaned to < 20 mg per day, and biologic agents and immunomodulators will be stopped at time of surgery. Prior to surgery, all patients must have had a colonoscopy with biopsy to confirm disease of the (neo)terminal ileum, and a recent update of imaging (for example ultrasound, MR or CT enterography). Baseline characteristics will be collected. Patients will all be treated in an enhanced recovery protocol.
SurgeryAt the time of surgery, all the procedures will be initiated as a laparoscopic procedure with conversion to an open operation only if clinically indicated. Close bowel ileocolic resection is advised to preserve vascularization.
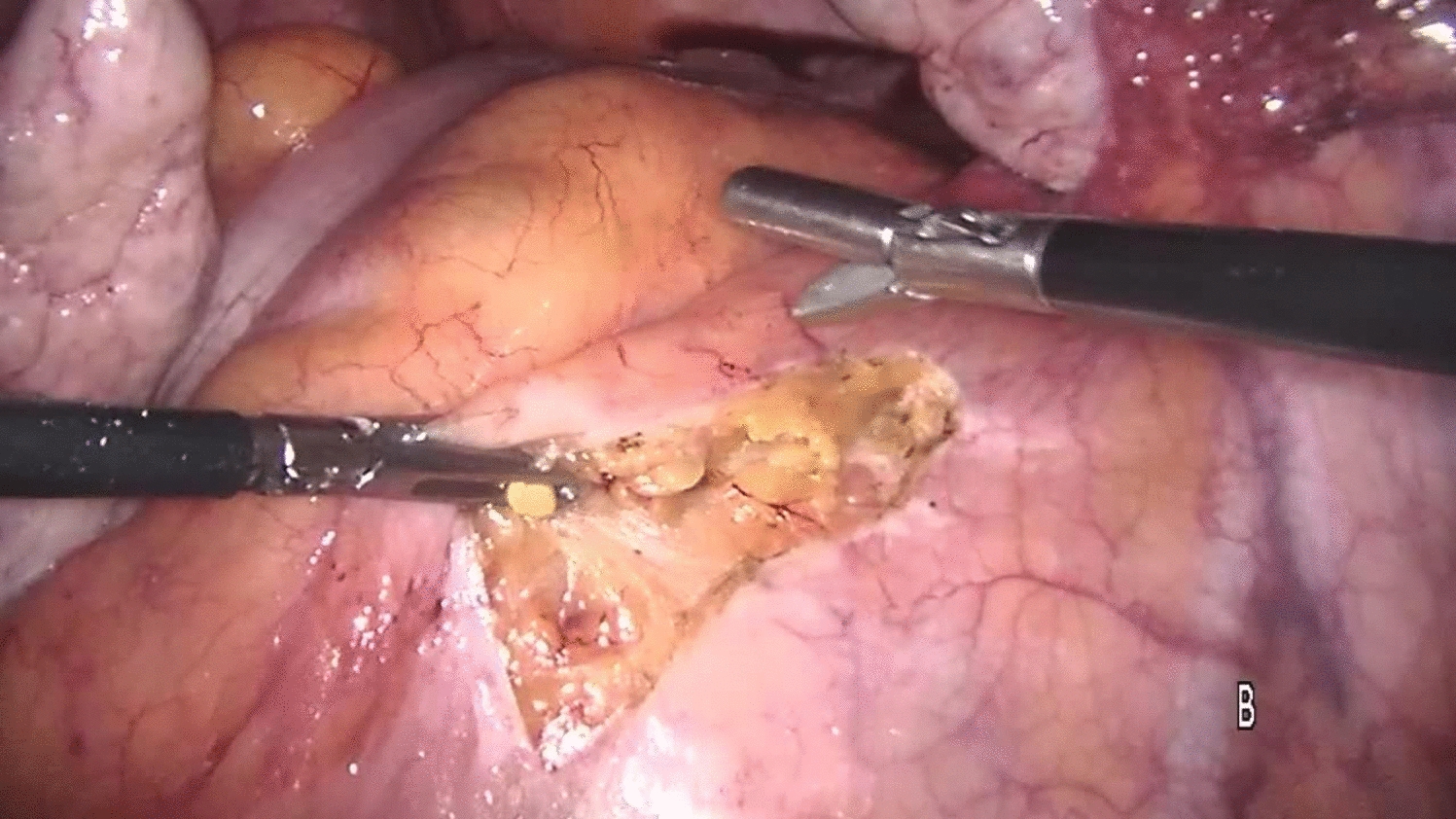
Group I: End-to-end handsewn or Kono-S anastomose. The end-to-end anastomosis is fashioned either by enlarging the small bowel diameter with an antimesenteric incision to fit the large bowel lumen or by tailored resection of a part of the staple line of the cross stapled colon. The Kono-S anastomosis is made according to the description by Kono [23]. The length of the actual anastomosis should be approximately 7 cm (Figs. 1, 2, video).
Group II: The side-to-side anastomosis is done according to local practice using a linear stapler either anisoperistaltic or isoperistaltic.
The rest of the operation will be identical. Care should be taken to ensure that, as a routine, the colon is anastomosed just beyond the ileocolic angle. Operative, postoperative and pathology data will be collected.
Quality control of surgical procedureTo ensure that the surgical technique is performed correctly in all centres, quality control will be conducted. A video vignette of the surgical procedure will be shared with all participating centres as an example (video). Furthermore, the participating centres will be asked to provide photos of the anastomoses and the resected specimen. Finally, each centre will provide videos of the procedure when requested.
Postoperative Postoperative morbidityData on postoperative morbidity will be collected. All adverse events (AEs) and severe adverse events (SAEs) will be documented throughout the hospital stay and outpatient visits.
Assessment of primary endpoint (endoscopic recurrence)All patients will undergo endoscopy with biopsies taken from the distal ileum and proximal colon on either side of the anastomosis to assess endoscopic and histologic recurrence at 6 months after surgery. Endoscopic results will be classified according to the SES-CD subdivided according to location and the (modified) Rutgeerts scoring system [24]. All colonoscopies must be video recorded according to a standard operational procedure (video). The quality of endoscopic scoring and over scoring will be assessed comparing the scoring of the local gastroenterologist with that of the central reading panel.
After endoscopic assessment at 6 months, medication can be initiated according to the severity of endoscopic recurrence according to the guidelines and the treating physician’s preference.
Assessment of clinical recurrence, quality of life and health care consumptionThe Crohn’s Disease Activity Index (CDAI) is determined at baseline and at 6 and 12 months postoperatively based on seven-days scoring by the patient before to these visits. Quality of life questionnaires are administered at each visit (preoperative, postoperative at 3 months, at 6 months and 12 months: the EuroQol, (EQ-5D-5L), 36-Item Short Form Health Survey (SF-36), and Inflammatory Bowel Disease Questionnaire (IBDQ). All types of healthcare consumption within the first year will be recorded, including readmission, emergency room visits, examinations, medical therapy etc.
Medical managementPatients will be treated according to the ECCO guidelines with respect to prophylactic medication after surgery.
Assessment of surgical recurrencePatients will be followed up for up to 5 years after surgery to determine the reoperation rate for recurrent disease at the anastomotic site.
Primary and secondary outcomesThe primary outcome is endoscopic recurrence at 6 months following ileocolic resection, defined as Rutgeerts score ≥ i2b assessed by local and central reading separately and blinded for the type of anastomosis. The quality of endoscopic scoring and over scoring will be assessed comparing the local scoring by the treating gastroenterologist with the central reading scoring for the different types of anastomosis.
Secondary outcomes include postoperative morbidity, the 6 months histologic and clinical recurrence rate and the need of restarting immunosuppressive medication within the first year postoperatively for endoscopic or clinical recurrence. For long term results, patient will be followed for at least 5 years to determine reoperation rate for recurrent disease at the anastomotic site. Additionally, quality of life will be measured with IBDQ, EQ-5D and SF- 36 together with health care consumption and costs.
Sample size calculationThe primary endpoint is the post-operative endoscopic recurrence of Crohn’s disease at 6 months.
The study is powered to detect a clinically relevant difference of 25% endoscopic recurrence at 6 months between the two randomized surgeries, 50% versus 25%. Assuming a test with continuity correction with 90% power, and an two-sided alpha-level of 0.05, in 2:1 ratio a total of 63 patients in each surgical arm will be required, for a total enrolment of 189 patients including an anticipated dropout of 10% in the Italian HAND2END study. In the Dutch End2End study, assuming a test with continuity correction with 80% power, and a two-sided alpha-level of 0.05, in 2:1 ratio a total of 55 patients per arm will be required, for a total enrolment of 165 patients, allowing for a 10% dropout.
After enrolling 189 patients in Italy, requiring 2.5 years with a 0.5-year follow-up, or 165 patients in the Netherlands, requiring 2 years with a 0.5-year follow-up, an interim analysis is done to determine whether the handsewn anastomoses are superior to stapled side-to-side anastomosis. If the interim analysis shows superiority of the handsewn anastomosis, the trial will continue with the end-to-end and the Kono-S anastomosis only. To find a reduction in endoscopic recurrence from 28 to 13% within the two handsewn groups (end-to-end and Kono-S), an additional 88 patients need to be included, for a total of 277 in the HAND2END. In Italy, a total 189 or 277 patients will be required over 3 or 4 years.. In the Dutch study, another 71 patients per group need to be included, leading to a total of 307 patients. In the End2End study, a total of 165 or 307 patients are needed over 2.5 or 4 years. If no superiority within the handsewn anastomosis is expected from the interim analysis, the study will be stopped after inclusion of either 189 patients in Italy or 165 patients in the Netherlands, plus those that are already included in the period needed to reach the 0.5-year follow-up.
The Dutch sample size has been adjusted according to the ZonMW first-round review committee’s preference, explaining why the the Dutch numbers are slightly different from those in the Italian study.
Statistical analysisDescriptive statistics will be used to report baseline patient and surgical variables. Baseline patient and procedure characteristics and perioperative outcome parameters are categorical, continuous and dichotomous variables, and will be presented accordingly. The chi-squared or Fisher’s exact test will be used to analyse the differences between the proportion of patients with recurrence.
Primary endpointDifferences in endoscopic recurrence at 6 months between the handsewn and stapled anastomosis groups will be analysed using logistic regression, which will be corrected for stratification factors and other potential confounders. Univariate associations with the risk of recurrence will be assessed using a Cox proportional hazards model, results will be reported as a hazard ratio (HR) and 95% confidence interval (CI).
Multiple variable models will be considered in the same way depending on the number of recurrences identified. The two-sided alpha-level will be set at 0.05 for statistical significance.
Differences in QoL will be analysed using mixed-model analysis of variance for repeated measures.
At the time of analysis, the most recent version of the statistical program IBM SPSS Statistics for Windows, Armonk, NY: IBM Corp. will be used.
The statistical analysis plan will be finalized before the data is locked for analysis. Decisions will be made regarding planned subgroup analyses and how to address protocol violations and any potential baseline imbalances.
Safety reportingThis study is considered a low-risk trial, because it involves the comparison of three well-established and commonly performed techniques in IBD surgery. No data and safety monitoring board (DSMB) will be installed. All adverse and serious adverse events (SAEs) will be monitored until they have abated or until a stable situation has been reached. The reporting procedure applies to all (S)AEs occurring up to 30 days after the surgical procedure and to any SAE that occurs after the 30-day period, if it is considered to have a reasonable possibility to be related to the protocol treatment or study participation. The study coordinator will report all SAEs to the accredited institutional review board that approved the study protocol.
Data handling and monitoringEach enrolled patient will be assigned a six-digit study number and all communication will occur using this number. The full name and date of birth of the patient will only be documented on the informed consent form, and these details will be securely kept in the participating hospital. In each of the participating hospitals, one surgeon acts as local investigator who is primarily responsible for execution of the study in compliance with the study protocol and ensuring the accuracy and completeness of the case report form (CRF). Data will be digitally collected and stored using the electronic data management system Castor EDC version 1.6 (www.castoredc.com). Continuous data monitoring will ensure complete and real-time prospective recording of data. Independent monitoring of the study progress and study quality will be performed by the Sponsors Clinical Monitoring Center (CMC).
Public disclosure and publication policyThe HAND2END and End2End trials are registered at www.ClinicalTrials.gov, with registration numbers rNCT05246917 and NCT05578235, respectively. The results of both studies will be submitted separately to peer-reviewed journals, regardless of study outcomes. Additionally, the results will be presented at international conferences and disseminated to relevant surgical and IBD associations. Co-authorship will be based on the International Committee of Medical Journal Editors (ICMJE) guidelines.
留言 (0)