Antibody
AR (Millipore Cat# 06-680, RRID:AB_310214), ARv7 (Precision antibody Cat# AG10008, RRID:AB_2631057), Bcl-2 (R&D Systems Cat# AF810, RRID:AB_355621; Cat# MAB8272, RRID:AB_10890789; Dako clone 124 Cat# M0887, RRID:AB_2064429), β4 integrin (Abcam, Cambridge, UK, #ab133682, RRID:AB_2923284, and 450-11 A, RRID:AB_396065, as in [24], β-actin (Abcam Cat# ab8227, RRID:AB_2305186), E-cadherin (GeneTex Cat# GTX100443, RRID:AB_10729586 and Abcam, #ab231303, RRID:AB_2923285), Cytokeratin (Agilent Cat# GA053, RRID:AB_2892089 and Santa Cruz Biotechnology Cat# sc-81,714, RRID:AB_2191222), Goat-anti-Mouse Alexa Fluor 546 (Thermo Fisher Scientific Cat# A-11,003, RRID:AB_2534071), Goat anti-Mouse IgG HRP (Bio-Rad Cat# 170–6516, RRID:AB_11125547), Goat-anti-Rabbit IgG Alexa Fluor 546 (Thermo Fisher Scientific Cat# A-11,010, RRID:AB_2534077), Goat-anti-Rabbit IgG HRP (SeraCare KPL Cat# 5220 − 0336, RRID:AB_2857917), IgG (Bethyl Cat# P120-101, RRID:AB_479829), JMJD3 (Abcam Cat# ab38113, RRID:AB_943898), H3K27me3 (Active Motif Cat# 39,155, RRID:AB_2561020), HSP90 (Cell Signaling Technology Cat# 4877, RRID:AB_2233307), UTX (Abcam Cat# ab36938, RRID:AB_883400).
Cell cultures, treatment, and transfection
PC-3M-luc2 were from Caliper Life,# 124,089 (RRID: CVCL_5J25) and provided by Prof. Carlo Leonetti (Istituto Nazione dei Tumori Regina Elena, Rome, Italy). 22Rv1-luc were generated from 22Rv1 (RRID: CVCL_1045) and kindly provided by Prof. Michael Henry (The University of Iowa, Iowa City, IA, USA) [41]. Western blot evaluated specific epithelial prostate markers, androgen receptors, full-length or ARv7 variant, and cytokeratin (Figure S1A). PC-3M-luc2 and 22Rv1-luc cells were grown in MEM (Corning, New York, USA, #15-010-CVR) and RPMI medium (1640 Corning, #10-040-CV), respectively, supplemented with 10% FBS (GIBCO, #10270106), 1% glutamine (Corning #25,005-CI), 1% penicillin and streptomycin (Corning #30002-CI). Medium for 22Rv1-luc cells was supplemented with 1% HEPES (Corning #25060-CI), 1% sodium pyruvate (Corning #25000-CIR), and 1% glucose. Cells were incubated at 37 °C with 5% CO2. GSK-J4 was prepared as described in [34]. Indirect (Hoechst) methods routinely screened all cell lines for mycoplasma contamination. The genetic identity of PC-3M-luc2 and 22Rv1-luc cell lines were authenticated by BMR Genomics (Padova, Italy) in October 2022. Cells were treated with GSK-J4 for the time and concentration indicated in the figure’s legend. Transient RNA interference for H19, KDM6A, and KDM6B was performed using TriFECTa Kit DsiRNA Duplex (Integrated DNA Technologies) according to the manufacturer’s instructions.
H19 silencing and overexpression
PC-3M-luc2 and 22Rv1-luc cells were stable engineered using recombinant GFP-expressing lentiviral vectors for H19 silencing (siH19, Origene#TL318197V) compared to scramble vector (Vector, Origene#TR30021) or H19 overexpression (oeH19; Genecopoeia#LPP-CS-GS1189L-Lv201-01-050) compared to empty vector (EV, Genecopoeia#LP146-050). Lentiviral infection was performed using 2.5 × 106 TU of lentivirus dissolved in 1 mL of medium supplemented with polybrene 8 µg/mL. 105 cells were plated in a 6-well plate one day before infection. GFP expression was evaluated after five days post-transduction (Figure S1B).
RNA extraction, cDNA preparation, real-time PCR, and droplet digital PCR
According to the manufacturer’s instructions, RNA from cells and tissues was extracted using Trizol (ThermoFisher). cDNA preparation and quantitative real-time PCR were performed as in [24] on QuantStudio 5 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using SYBR Green quantification. The relative amount of each gene was measured as 2−ΔCt. β-Actin or P0 served as endogenous control. cDNA preparation, preamp PCR, and digital droplet PCR (ddPCR) were performed on QX200 droplet digital PCR system (Biorad) as described in [42]. Primers to H19, CDH1, ITGB4, P0, GAPDH, and β-actin as in [24], PSA as in [43]. Primers were as follows:
hKDM6A 5’-CAGTTAGCTTTGGTTGACTGTAATCC-3’ and 5’- AGTGGGCAATGTGAAATTGAATT-3’;
hKDM6B 5’-TGTGGAACTTGCTACACCTTGAG-3’ and 5’-GGTTCAGCAGCTCGCTTCAC-3’;
Protein extraction and western blotting
Proteins were extracted and prepared using Trizol (ThermoFisher) as described in [24]. Western blots were performed using 15–20 µg of protein extract resolved by 4−12% gradient Invitrogen Precast gel (MES buffer) and revealed with an ECL Western Blot Detection Kit (Amersham Pharmacia Biotech, Buckinghamshire, England) using UVIDOC (Eppendorf S.r.l., Hamburg, Germany). Densitometry analysis was performed with ImageJ software (version 1.8.0).
Immunofluorescence and confocal microscopy
Confocal analysis was performed as described in [44]. Briefly, cells were fixed in 4% paraformaldehyde and incubated with anti-E-cadherin antibody (Abcam, #ab231303) or β4 integrin (Abcam, #ab133682), diluted 1:250 in PBS 5% Goat Serum. Nuclei were counterstained with DAPI. Slides were analyzed with a confocal laser scanning system (Nikon Eclipse Ti2 confocal microscope) and Z stack images were processed by NIS Elements AR 5.30 software (Nikon Europe BV).
Proliferation assay
Proliferation was assessed using the IncuCyte system S3 Kinetic Live Cell Imaging System (Essen BioScience, Ann Arbor, MI, USA). PC-3M-luc2 and 22Rv1-luc cells were seeded at 8000 and 15000 cells/well, respectively, in triplicate on a 24-well plate (Corning#3524), with IncuCyte readings taken at six h-cycles starting from day 0 (9 or 16 images per well). Pictures were taken every 2 or 4 h, and the IncuCyte algorithm was used for phase area confluence calculations.
Trans-well invasion assays
Invasion assays were performed in triplicate as described in [24] with some modifications. Briefly, after 24 h in serum-free media, 1 × 105 cells were seeded in top chambers of 24-well transwell plates (Corning #3422) in FBS-free media with membrane inserts with matrigel coated. Invading cells were stained with 0.1% crystal violet. Images were obtained by AXIO microscope with AxioCam Erc5s, Zeiss, under 20X or 10X magnification.
Chromatin immunoprecipitation (ChIP)
ChIPs were performed as in [24], and analysis of DNA fragments was performed in duplicate by qPCR on QuantStudio 5 Real-Time PCR System (Applied Biosystems) using SYBR Master mix (Applied Biosystems, Foster City, CA, USA) with the evaluation of dissociation curves. Standard curves were generated by serially diluting the input (5-log dilutions in triplicate). The specific sequences isolated by the immune-complexes were normalized to the corresponding DNA input control, and data represented as relative enrichment. Immunoprecipitations were performed using specific antibodies to JMJD3 (Abcam, #ab38113), UTX (Abcam, #ab36938), and H3K27me3 (Active Motif #39,155, Carlsbad, CA, USA). IgG (Bethyl. #P120-101, Montgomery, TX, USA) was used as negative control. Primers for hCDH1 promoter were as in [24]. Primers for ITGB4 promoter were as follows:
hITGB4prom 5’- CTGGCCTGACACACACAGATCT-3’ and 5’-TTTGGGAACAATGTGGAAGGA-3’.
Animal
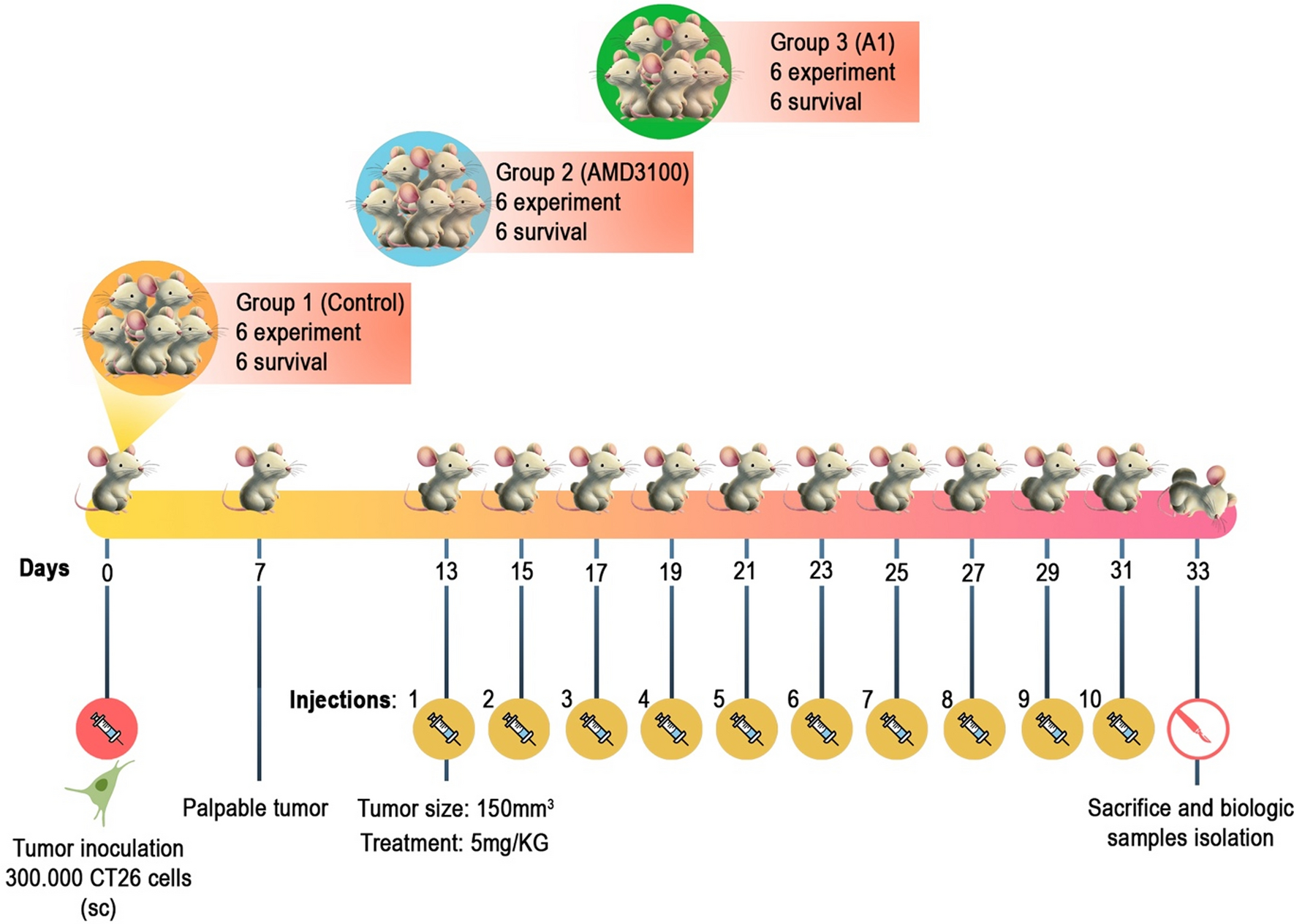
NOD/SCID (RRID: IMSR_JAX:001303) and NSG (RRID: IMSR_JAX:005557) mice from Charles River Laboratories were housed 3–4 for cage in a room with controlled temperature, constant humidity, and a 12 h light/dark cycle with free access to food and water. The standard xenograft mouse model was generated using 5-6-week-old male NOD-SCID mice subcutaneously injected with 3 × 106 cells/mouse with matrigel (1:1). Tumor growth was monitored by bioluminescence imaging (IVIS II Lumina, PerkinElmer Italy S.p.A., Milan, Italy). Data were acquired and analyzed using the living image software version 4.7.4 (Caliper Life Sciences). Tumor growth was measured with digital calipers, and tumor volumes were calculated from the formula V = (W2 x L)/2, W = width, L = length. In addition, a metastatic mouse model was generated by injecting 1 × 106 cells into the lateral tail vein of NSG mice (5–6 weeks old). Mice were randomly divided into two groups and treated with GSK-J4- (50 mg/Kg diluted 10:90 DMSO:20% captisol as in [45] and administrated via IP 5 days/week) or vehicle-treated (DMSO) starting from day 0. At the end of the experiments, subcutaneous tumors were collected for further analysis, and metastases on different organs were visualized and quantified by ex vivo bioluminescence using IVIS Lumina.
Histological analysis
For histological and immunohistochemical analyses, tissues were fixed in 10% formalin (Thermo Fisher Scientific). Unstained tissue Sect. (4 μm-thick) were cut from formalin-fixed, paraffin-embedded blocks and mounted on a positively charged glass slide. IHC was performed using the Leica Bond (LBO, Milan, Italy) immunostainer or a Ventana Benchmark XT automated immunostainer (Ventana Medical Systems, Tucson, AZ, USA) as follows: cytocheratin AE1/AE3, Agilent DAKO, #GA053, ER pH6 for 20 min. Hematoxylin was used for nuclear counterstaining. Whole slide imaging (WSI) was performed using NanoZoomer 2.ORS (Hamamatsu photonics, Hamamatsu, Japan) using 20X magnification (0.46 microns/pixel).
Organotypic slice cultures (OSCs)
PCa patients (n = 25) were enrolled at the Urology of Università Cattolica (Rome, Italy) to perform prostatectomy with the following inclusion criteria: (i) clinically localized PCa at diagnosis and (ii) absence of hormone treatment/radiotherapy before surgery. Of note, all OSCs were checked by the pathologist on the original histopathological slide for morphology, tissue architecture, and amount of tumor (≥ 75%). OSCs were generated as previously described in [24, 43, 46]. Briefly, medium was replaced daily and collected for cell death assay (see paragraph below). Slices were treated with GSK-J4 (5µM) for 72 h, and RNA/protein was extracted and analyzed as previously described [24].
Cell death assay
Apoptosis was determined with Cell Death Detection ELISA PLUS kit (Roche, Basel, Switzerland) using 5–20 µl extracellular-medium according to manufacturers’ instructions. Absorbance at 405 and 490 nm was assessed at VICTOR X4 (Perkin Elmer).
Statistical analysis
Data were expressed as mean ± SEM or as fold change as indicated in figure legends. Difference among ≥ 3 groups were analyzed with a Kruskal-Wallis test, and post hoc comparison was done using the Mann-Whitney U test (α = 0.05). Difference among 2 groups were analyzed with Mann-Whitney U test. Chi-square test were used to compare the proportion of the number of mets/mouse between groups as indicated in figure legend. Statistical analysis was performed using GraphPad Prism 8.0.2 statistical software. P-values of < 0.05 were considered significant.


留言 (0)