
記住我


The recombinant uricase enzyme was purified using previous methods, and the SDS-polyacrylamide gel electrophoresis technique was used to analyze the proteins eluted from the purification [40]. The observation of a single band with a molecular weight in the region of 35 kDa served as conclusive evidence for the expression of recombinant urate oxidase enzyme (Fig. 1) [41].
Fig. 1
Recombinant urate oxidase purification. proteins eluted collected after Ni-NTA chromatography were visualized on SDS-PAGE.
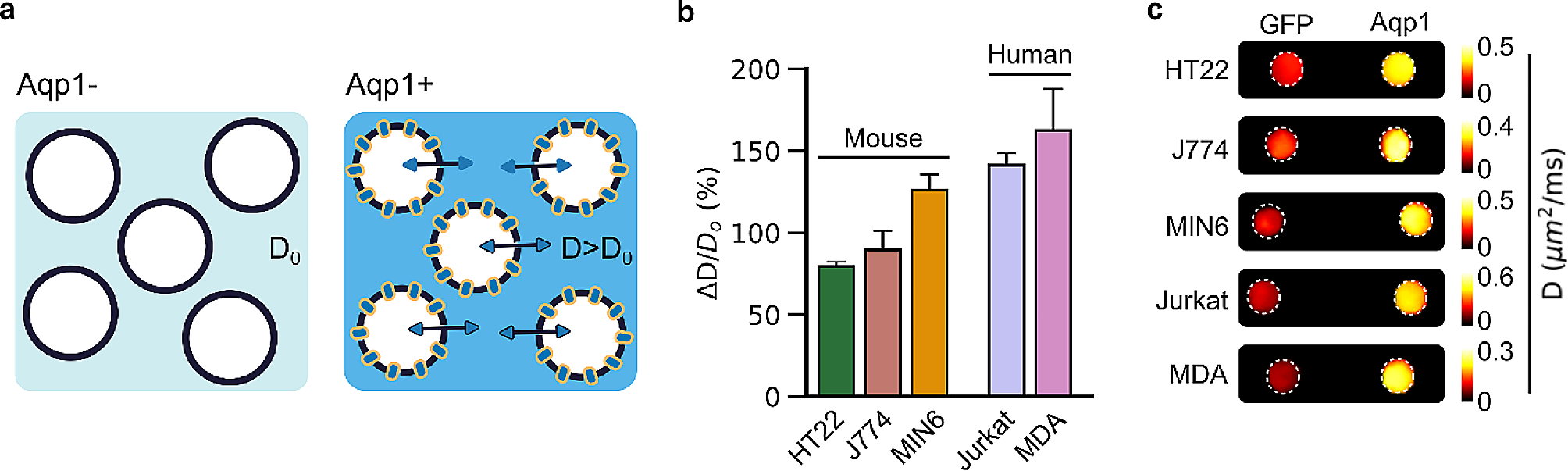
Optimum temperature and incubation time plus taurine concentration by RSMIn order optimize the three most prominent factors (temperatures, incubation time, and the concentration of taurine), a total of 18 full factorial central composite designs (CCDs) were used in this study, using Design-Expert software version 11. The predicted and experimental results are presented in Table 1.
Table 1 Design matrix of experiments proposed by response surface methodology (RSM) and the experimental units acquired for each experimentThe results shown in Table 1 demonstrate a high degree of concordance between the predicted response values and the corresponding experimental data. The RSM model suggested a quadratic polynomial equation. The ultimate equation, expressed in terms of actual The RSM model suggested a quadratic polynomial equation. The final equation, in terms of the actual factor, can be stated as follows:
$$\mathbf=+5.20717-0.049496\,\text-0.006595\,\text-0.151224\,\text-0.000152\,\text\,\ast\,\text+0.002154\,\text\,\ast\,\text+0.000293\,\text\,\ast\,\text+0.000557\,\text^2+\text\,\text^2-\,\text\,\text^2$$
The presence of a positive coefficient indicates a synergistic effect on the response, while a negative coefficient signifies an antagonistic effect. The RSM plot in Fig. 2 illustrates the impact of taurine concentration, incubation time, and temperature on the activity of the recombinant uricase enzyme.
Fig. 2
The planned set of contour plots aims to illustrate the impact of three independent variables and their effects on response (activity): (a1/2) The influence of temperature, taurine concentration, and their interaction on activity, while keeping the incubation time constant at 25 min (b1/2) The impact of temperature, incubation time, and their interaction on activity, while maintaining a constant taurine concentration of 250 mM. (c1/2) The effects of taurine concentration, incubation time, and their interaction on activity, while holding the temperature constant at 28 °C
Based on the results shown in Fig. 2a1/2, it can be observed that at high temperatures with increasing taurine osmolyte concentration and at low temperatures with lower taurine osmolyte concentration, the enzyme achieves its highest possible level of activity. Moreover, it is evident from the data presented in Fig. 2b1/2 that the enzymatic activity increases at higher temperatures with increases in the incubation time of treated uricase enzyme with a constant concentration of taurine osmolyte. Moreover, Fig. 2c1/2 indicates that both a low taurine concentration and a long incubation time had a positive effect on the activity of the recombinant uricase enzyme, and a high taurine concentration and a short incubation time had the same effect. These results suggest that the effects of all three variables and their interactions with each other on the recombinant uricase enzyme activity have been significant. The optimum conditions for achieving maximum activity of the recombinant uricase enzyme were determined to be a temperature of 28 °C, a taurine concentration of 450 mM, and an incubation time of 25 min. The recombinant uricase enzyme activity of 2.05 U/ml was generally achieved under optimal conditions, as indicated in Table 1.
Thermal inactivation and stabilityTo investigate the mechanism of protein inactivation, it is crucial to assess kinetic and thermodynamic parameters, which are fundamental in thermal processes. The study investigated the irreversible thermal inactivation of enzymes that were treated with taurine and enzymes that were not treated with taurine, at various temperatures. In general, a population of protein molecules undergo irreversible structural changes, which can be seen from the reduction of the residual activity of the pristine enzyme at representative temperatures. Thus, a resetting of the operating temperature does not lead to the reestablishment of the initial enzyme activity. The time and temperature of incubation, as well as the specific protein type, determine the degree of reversibility for each protein. The examination of the thermo-inactivation process following a duration of 60 min indicated that the untreated enzyme exhibited lower stability compared to the enzyme subjected to taurine treatment across the temperature range of 40, 55, and 60 ˚C, particularly at elevated temperatures. The enzyme’s activity was shown to retain 40% and 20% of its initial activity after being incubated for 60 min at 55 °C with and without the presence of taurine, respectively (Fig. 3). After incubating the recombinant uricase enzyme with and without taurine osmolyte at representative temperatures within 40, 55, and 60 ˚C, irreversible thermal inactivation experiments were performed for 1 h for detailed analysis. The results indicated that during a 60 min incubation at 40 ˚C, the enzymes treated with taurine exhibited a retention of over 70% of their activity, as depicted in Fig. 4a, b, and c. The variation in melting points of proteins can be attributed to the distinctive primary sequence of amino acids that are characteristic of certain protein types. Various factors, such as pH and salt concentration, as well as post-translational modifications, can significantly impact the stability of protein structure and hence affect the melting temperature. Protein stability increases proportionally with greater values of the thermal melting temperature (Tm). In order to achieve the minimum level of activity, Amin, the data of treated and untreated uricase enzyme were plotted until no further drop in enzyme activity (ARes/A0) was observed (Fig. 5a/b). Then, according to Fig. 5c/d, the first parameter of thermal stability, i.e. the melting temperature of the treated and untreated uricase enzyme (Tm) was estimated.
Fig. 3
Irreversible thermal inactivation of taurine treated enzyme (white) and pristine enzyme (black) at different temperatures for 60 min
Fig. 4
Irreversible thermal inactivation of taurine treated enzyme a 40 °C, b 55 °C, and c 60 °C for different time intervals
Fig. 5
Determination of thermostability parameters: a and b, Reduction in the proportion of ARes compared with native activity (A0) over time in treated and untreated enzyme. Amin is the minimum activity at a given temperature that corresponds to the enzyme in the equilibrium state which represents the total amount of active enzyme upon cooling a the treated enzyme b the untreated enzyme. b and c, Estimation of melting temperature (Tm) that corresponds to the temperature when Amin drops down to 50% of the native activity (A0) in treated and untreated enzymes respectively.
Kinetic parametersBased on previous kinetic studies, the Michaelis constant (Km) values of the urate oxidase enzyme were investigated under two conditions: in the absence and presence of taurine. Table 2 presents a comprehensive summary of the kinetic parameters acquired in the present study. The taurine-treated enzyme exhibited a twofold augmentation in Km compared to the untreated uricase enzyme. Based on the results, it can be shown that the enzyme treated with taurine osmolyte exhibited a greater affinity towards the substrate in comparison to the untreated uricase enzyme (as depicted in Fig. 6a, b, and Table 2). A high Km value indicates that a substantial amount of substrate is required to achieve enzyme saturation, implying that the enzyme exhibits a low affinity for the substrate. Conversely, a low Km value signifies a minimal quantity of substrate required to achieve enzyme saturation, indicating a higher affinity for the substrate. Hence, the data indicate that there is a change in the rate of conformational change in the presence and absence of taurine on the way to forming the Michaelis complex. An increase in the Km value of the enzyme treated with taurine may indicate that the enzyme’s structure underwent alterations during the stabilization process or that taurine introduces a spatial barrier that hinders the entry of the substrate (uric acid) into the enzyme’s active site [21, 24, 42, 43]. Furthermore, the enzyme’s catalytic efficiency or rate constants for the catalytic conversion of substrate into product (Kcat) and the ratio of Kcat to Km, which serves as an indicator of the specificity of the enzyme to the substrate, were examined in both the absence and presence of taurine osmolyte. The observed reduction in the value of this parameter for the enzyme in the presence of taurine osmolyte is probably attributable to the spatial barrier.
Fig. 6
The kinetic parameters of uricase in the absence (a) and presence of taurine (b)
Table 2 The biochemical characteristics of uricase in the absence and presence of taurineFluorescence measurementsFluorescence spectroscopyThe utilization of fluorescence spectroscopy has been widely employed to investigate the interactions between ligands and proteins, yielding valuable insights into the quenching mechanism, binding constants, and binding sites [44, 45]. Emission spectra alterations can yield insights about the structure and dynamics of a molecule [46, 47]. The main source of fluorescence in the uricase enzyme is attributed to the presence of tryptophan (Trp) and tyrosine (Tyr) residues. The Trp and Tyr residues of proteins have maximal fluorescence intensities of approximately 340 nm and 300 nm, respectively, when stimulated at 280 nm. Figure 7a, b, and c illustrates the fluorescence spectra of the uricase enzyme at different taurine concentrations. The fluorescence intensity of uricase showed a consistent reduction as the concentration of taurine increased, suggesting that taurine interacted with uricase and influenced its structure [48, 49].
Fig. 7
Emission spectra of uricase in the presence of various taurine concentrations of (λexc = 280 nm) at a: 40 ˚C, b: 55 ˚C and c: 60 ˚C
Fluorescence quenching mechanismFluorescence quenching refers to the reduction in the efficiency of fluorescence emission by a fluorophore due to various chemical interactions, including excited-state reactions, formation of ground-state complexes, energy transfer, and collisional quenching. The methods of quenching can be categorized as either dynamic quenching or static quenching. These categories are differentiated by their respective dependencies on temperature and viscosity, or preferable by measuring their lifetimes. As the temperature increases, the diffusion coefficients also increase. Consequently, the dynamic quenching constants will also increase with rising temperature. In contrast, the rise in temperature is expected to lead to a reduction in the stability of complexes. Therefore, it is anticipated that the values of the static quenching constants will be lower [50]. The well-known Stern–Volmer equation was used to confirm the mechanism as follows [51]:
$$\frac_}=1+_}_\left[Q\right]=1+_\left[Q\right]$$
F0 and F denote the fluorescence intensity when there is no quencher and when the quencher is present, respectively; [52] represents the concentration of the quencher, while τ0 denotes the fluorescence lifetime when there is no quencher present. The value of τ0 is always 10−8 seconds [53]. Kq represents the rate at which the biological macromolecule is quenched, whereas KSV denotes the Stern-Volmer quenching constant. KSV is also denoted by the following equation:
This study aimed to investigate the quenching mechanism of uricase by obtaining fluorescence quenching spectra at three distinct temperatures (40, 55, and 60 °C) in the presence of various taurine concentrations. Figure 8 displays the Stern-Volmer plots depicting the quenching of uricase fluorescence by taurine. The computed KSV and Kq values are presented in Table 3. A linear Stern-Volmer plot typically suggests the presence of a single class of fluorophore, which is equally susceptible to quenching by the quencher. The assessment of static and dynamic quenching can be determined by observing the impact of temperature and viscosity. Another, more preferable approach is to conduct studies on fluorescence lifetime [22]. Increasing temperatures cause diffusion to occur more quickly, leading to an increase in collisional quenching. Additionally, higher temperatures typically cause loosely bound complexes to separate, resulting in a decrease in static quenching. Dynamic quenching is indicated by the rise in KSV value with increasing temperature. The Kq value was < 2.0 × 1010 L mol−1 s−1. Furthermore, the observation suggests that the decrease in uricase fluorescence caused by taurine is a form of static quenching, as seen in Fig. 7a, b, and c. The impact of dynamic quenching is widely acknowledged to affect only the excited state of the fluorophore, whereas static quenching alters the absorption spectrum of the fluorophore. This study showed that the quenching process occurs through static quenching, which is triggered by the formation of the uricase-taurine complex in its ground state. Osmolytes tend to increase the stability of proteins’ natural structure. In conclusion, these findings suggest that the interaction between the protein and the quencher is a complex one, which is more consistent with the static quenching process rather than dynamic collision quenching.
Fig. 8
Stern–Volmer plots for the quenching of uricase by taurine at 40, 55, and 60 ˚C
Table 3 Stern-Volmer constants for the interaction of uricase with taurine at different temperaturesCalculation of binding parametersThe equation provided can be used to compute the binding constant (Kb) and the number of binding sites (n) in the static quenching process, where small molecules independently bind to a group of identical sites on a macromolecule.
$$\text\left(\frac_-F}\right)=log_+nlog\left[Q\right]$$
where [52] is the concentration of the quencher; F0 and F are the fluorescence intensity in the absence and presence of the quencher, respectively; Ka is the binding constant, and n is the number of binding sites per uricase molecule. By plotting the logarithm of (F0 − F)/F against the logarithm of [52] (as depicted in Fig. 9), one may determine the values of n and Ka [54]. The results are succinctly presented in Table 4.
Fig. 9
Double-log plots of the taurine quenching effect on uricase enzyme fluorescence at different temperatures
Significantly, the values of n at the experimental temperatures were nearly identical to 1, suggesting that there is just one binding site in uricase for taurine. Furthermore, the increase in temperature resulted in a decrease in the Ka value, indicating a decrease in the stability of the uricase-taurine complex. Therefore, the binding process was characterized as an exothermic reaction.
Table 4 Binging and thermodynamic parameters of UOX-Taurine interaction at three different temperatures Thermodynamic parameters determinationThe binding constant is influenced by temperature, indicating that the thermodynamic process is responsible for the creation of the uricase-taurine complex. An analysis of this dependency was made to reveal the interacting forces between taurine and uricase. Four non-covalent-type interactions attribute a major part of the bonding between ligands to themselves. The hydrophobic force, van der Waals forces, hydrogen bonding, and electrostatic forces are some of these interactions [22]. The major forces were determined based on the sign and magnitude of the thermodynamic parameters. The Vant Hoff equation can be used to approximate the value of ΔH and ΔS if they are only slightly variable and do not change much over the studied temperature range.
$$\beginLn_=-\frac+\frac\\\varDelta G=-RTLn_=\varDelta H-T\varDelta S\end$$
where Ka and T refer to the binding constant and temperature, respectively, and R is the gas constant (8.314 J mol−1 K−1). The ΔG values were determined and the obtained results were shown in Table 4. Also, the ΔH and ΔS values were calculated by graphing \(Ln_\) vs. \(\frac\) (Fig. 10).
Fig. 10
plot lnKa against 1/T for interaction taurine with uricase at 313, 328, and 333 K
Ross and Subramanian have provided a comprehensive summary of the forces that govern the interactions between proteins and ligands, allowing for the identification of many forms of binding with distinct interactions. If the enthalpy change (ΔH) is negative and the entropy change (ΔS) is negative, then the binding reaction is mostly governed by van der Waals forces and hydrogen bond interactions. When the change in enthalpy (ΔH) is greater than zero and the change in entropy (ΔS) is also greater than zero, it indicates that hydrophobic interactions are the prevailing factor. If the change in enthalpy (ΔH) is negative and the change in entropy (ΔS) is positive, then the primary driving factors are electrostatic interactions. Enthalpy change (ΔH) can be considered constant when the temperature range is minimal. The utilization of enthalpy change (ΔH) and entropy change (ΔS) can serve to validate the binding modalities [55]. The interaction and forces governing the relationship between taurine and uricase can be described as follows: The negative values of ΔS and ΔH indicate that the van der Waals force and the hydrogen bonding interactions were the main factors influencing in the interaction of taurine with UOX. Furthermore, the negative ΔG values suggest that the binding process occurred spontaneously.
ANS fluorescenceThe fluorescent probe ANS is employed for the purpose of identifying protein binding sites. The present investigation involved the utilization of spectroscopy techniques under controlled conditions, specifically at a temperature of 28 °C. The excitation wavelength employed was 370 nm, whereas the emission wavelength range spanned from 400 to 600 nm. Figure 11 displays the fluorescence emission curves pertaining to the interaction between ANS and both treated and untreated uricase enzymes. After the binding of ANS to the hydrophobic surface of the treated enzyme, there was a significant increase in fluorescence intensity. The results suggest that there has been a modification in the composition and conformation of the enzyme’s tertiary and quaternary structures, which has led to the placement of hydrophobic residues on the enzyme surface. This may allow for the establishment and arrangement of more stable hydrophobic regions or clefts by increasingly shielding them from a polar environment, thereby driving equilibrium towards the formation of a more stable protein complex [23, 56,57,58,59,60].
Fig. 11
Fluorescence study of uricase in the absence and presence of taurine. Extrinsic fluorescence measurement of uricase using ANS. ANS (dashed line), ANS along with free uricase (dotted line), along with taurine treated uricase (solid line); The spectrums were taken by excitation at 280 nm
Molecular dynamics simulation resultsMD simulations can be a valuable mean to gaining insight into the conformational properties of the enzyme [40, 61]. Our obtained theoretical results are valuable to improve the stability and performance development of UOX. MD simulations for native and taurine-treated enzymes were done through MD simulation package GROMACS 5.1.4 which adopts the CHARRM27 force field parameter for energy. To understand the enzyme activity, it is necessary to examine the protein structure in the presence of taurine.
RMSD and secondary structure analysisThe root mean square deviation (RMSD) and the root mean square fluctuation (RMSF) of Cα atoms of two simulated enzymes were determined and illustrated in Fig. 12a and b.
Fig. 12
The root mean square deviation of Cα atoms (Cα-RMSD) and the root mean square fluctuation of Cα atoms (Cα-RMSF) of uricase in the absence (blue line) and presence of taurine (pink line)
The RMSD values reached saturation very fast, after about 2.3 ns, after which the variations reached the minimum values (see Fig. 12a). This evidence suggests that each system is simulated for a sufficient time to reach equilibrium and that the simulation time is enough to cover all possible states. At the end of the simulation, the RMSD values of naked and treated enzymes reached ~ 0.310 nm and ~ 0.144 nm, respectively. The lower Cα-RMSD value of the enzyme in the presence of taurine compared to the free enzyme indicates the stability of the UOX structure in the binary system.
The structural flexibility of both the naked and taurine-treated enzymes was assessed by calculating the Cα-RMSF (refer to Fig. 12b). In general, the pure water system exhibited greater fluctuations in residues compared to the binary system. Regions with higher flexibility were characterized by higher RMSF values. Consequently, the lower RMSF values observed in the binary system confirm that treating UOX with taurine enhances the stability of the enzyme.
The information about the local secondary structure changes has been obtained from the MD simulation. The UOX secondary structure during the 40 ns of MD simulations was investigated to examine the changes in the content of α-helix and β-sheet structures in the enzyme (See Fig. 13a and b).
Fig. 13
Secondary structure assignment of the protein as a function of time for a: UOX and b: UOX/taurine for 40 ns
According to these results, the higher β-sheet and α-helix content of taurine-treated enzyme suggest that taurine may be responsible for the stability and higher activity of the enzyme at elevated temperatures and may preserve the enzyme’s active site integrity.
Table 5 reports the percentage of the secondary structures for the free and treated enzymes during the MD simulation.
Table 5 Secondary structure of UOX in the two simulated systemsThe α-helix and β-sheets contents of the taurine-UOX enzyme (25.12% and 37.46%, respectively) increased compared with those of the free enzyme (23.90% and 32.61%, respectively). Further, the obtained results indicate that the β-bridge percentage in the treated enzyme has been higher than that of the naked enzyme (see Table 5). The obtained MD simulation results provide more details about the taurine effects on the enzyme structure.
The enzyme stability is estimated by a number of interactions including hydrogen bonding, electrostatic, hydrophobic interactions, and van der Waals forces [62, 63]. Thus, the taurine-UOX interaction changes the secondary structure composition of the enzyme. The hydrogen bond between the carbonyl oxygen and the amide hydrogen atoms forms the α-helix and β-sheet structures; thus, changes in the secondary structure composition of UOX may enhances its intramolecular H-bonds so that the content of the α-helix and β-sheet increases. In general, the results showed that taurine causes changes in the composition of the enzyme and thus boosts the enzyme activity, which is consistent with the results of the experimental section. The research findings confirmed the significant impact of taurine on the UOX stability.
In Fig. 14, it can be observed that the enzyme’s conformation was changed from a random coil to a β-sheet and α-helix in the binary system. This change in conformation could potentially maintain the integrity of the enzyme’s active site, contributing to its increased activity and stability at elevated temperatures.
Fig. 14
The structures of a: free UOX and b: treated-taurine UOX at the end of 50 ns simulation (active site residues have been indicated with red color)
SASA and hydrogen bondingSolvent accessible surface area (SASA) is a significant parameter to analyze the interactions between protein and solvent [
留言 (0)