
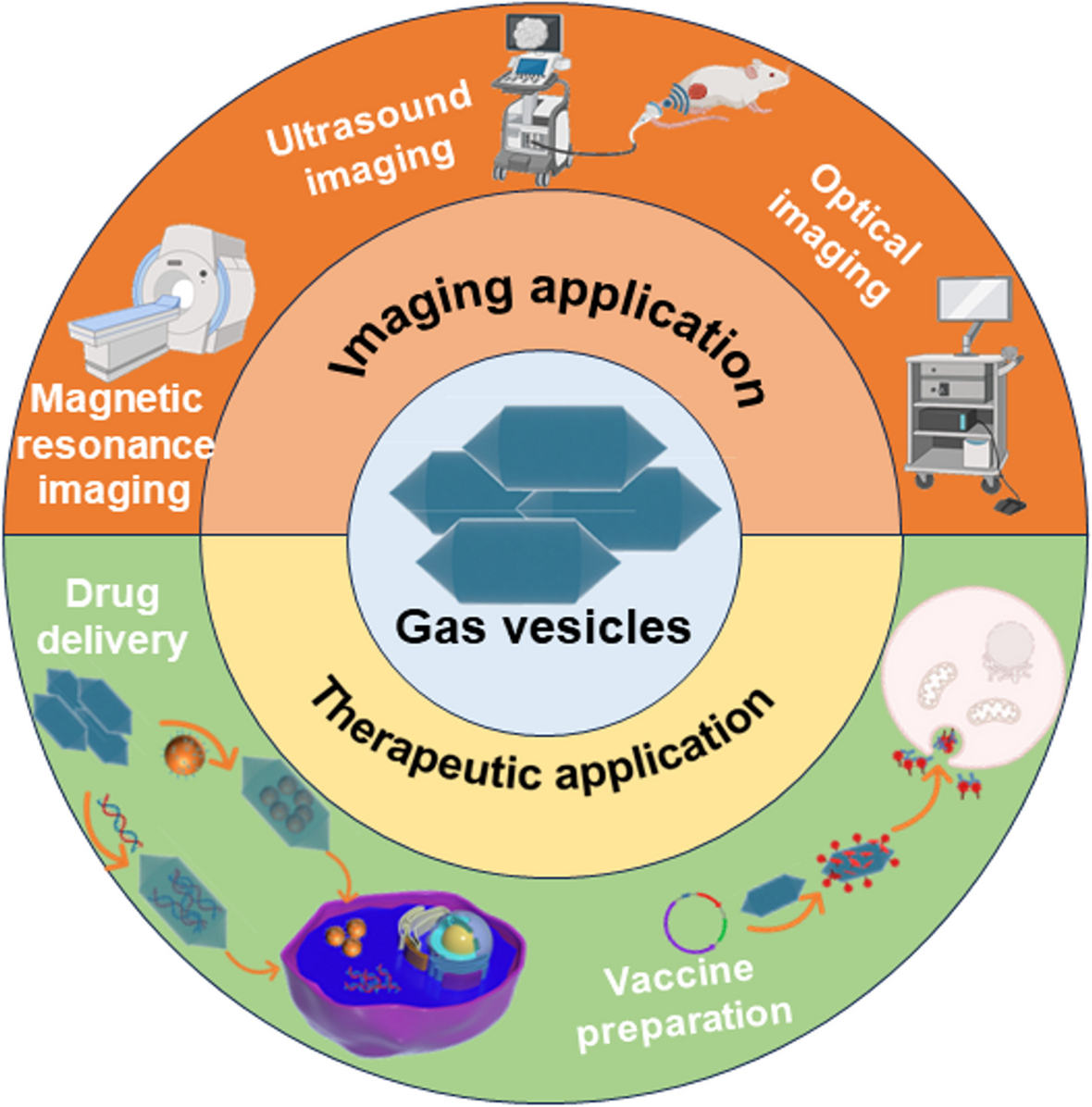
記住我
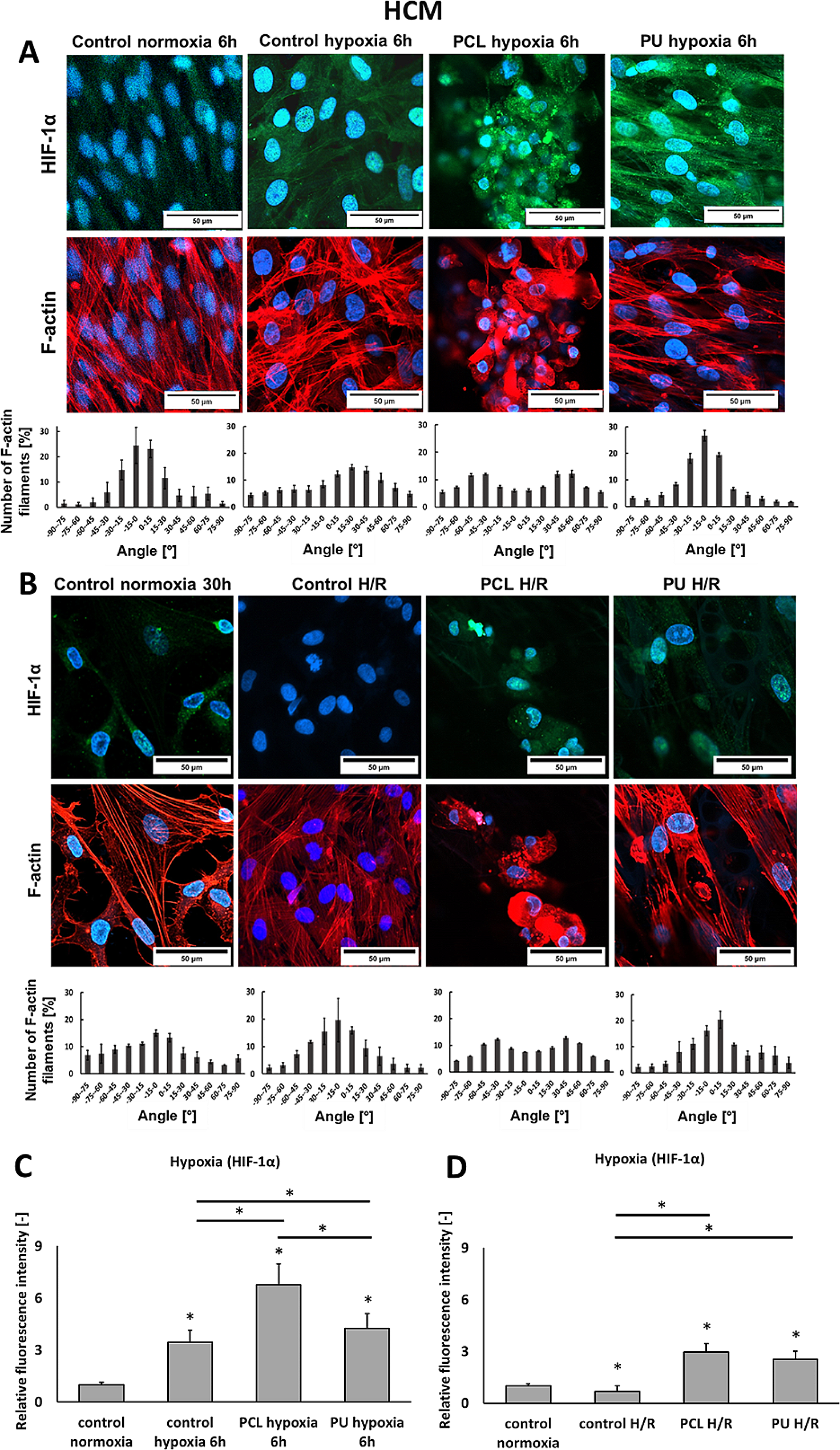
To identify suitable candidates for the construction of transcriptional reporters, FPs covering the entire spectra of visible light were evaluated (Fig. 1A). For this, 15 individual FPs were cloned into a plasmid downstream of the frequently used constitutive promoter (PrpsM) of the ribosomal protein S13 and introduced into S. enterica [15]. Subsequently, the excitation and emission spectra of the FPs in vivo were recorded using a plate reader (Table S1 and Figure S1) and resembled previous in vitro measurements available in FPbase (https://www.fpbase.org/) (Table 1) [15]. Next, we performed endpoint measurements to compare the fluorescence intensities for all constructs over wild-type (WT) background levels normalized by optical density (Fig. 1A). From this, we calculated the signal to background ratios and confirmed previous findings that mNeonGreen and mScarlet-I show a particularly high signal over background ratio, with a 1,000-fold increase in fluorescence over WT levels. Blue fluorescent proteins such as mCerulean depicted a 100-fold increase in signal to background, whereas sfmTurquoise2ox yielded only a 10-fold increase. In the green/yellow spectrum, mGFPmut2, Ypet, mVenusNB, and mNeonGreen show a 10-fold higher fluorescence intensity compared to other FPs within the same emission range (Fig. 1B). Bacteria exhibit only weak autofluorescence at wavelengths above 500 nm (Fig. 1A), which explains the relative high signal over WT background of FPs such as mCherry and mScarlet-I.
Fig. 1
Endpoint measurements of the fluorescent proteins evaluated in this study. A Relative fluorescent units normalized by optical density (OD600) are shown for cells constitutively (PrpsM) expressing the corresponding FP (colored bars), compared to the autofluorescence of S. enterica WT cells (white bars). The excitation and emission wavelengths for each FP are depicted in Table 1. Bar graphs represent mean values and standard deviations of at least three biological replicates. For evaluation of the spectra (Fig. S1) and endpoint measurements, cells were grown to exponential phase and fluorescence was directly measured in growth medium within a final volume of 200 µl in 96 well plates using a Synergy H1 plate reader. B Fold-change of reporter signal normalized to WT background fluorescence depicted for each FP
One major advantage of plasmid-based reporters is the ease of transforming a single construct into various backgrounds. However, plasmid-based reporters may skew the activity of a GOI because of enhanced gene dose due to copy number of the plasmid backbone [34]. Plasmid replication and reporter protein expression might also affect bacterial growth due the increased metabolic burden [35]. We addressed this, by comparing growth kinetics on population level and observed a slight delay of strains expressing plasmid based eGFP or mScarlet-I to reach stationary phase (Figure S2). Lastly, plasmids may be non-homogenously distributed during exponential growth, when daughter cells receive different number of plasmid copies, rendering observations of phenotypic heterogeneity more difficult [34]. To overcome these limitations, native or ectopic transcriptional fusions are preferable despite a reduction in signal strength due to decreased gene doses. Accordingly, we next conducted further analyses using chromosomal constructs. For this, we chose representative green FPs (mNeonGreen and eGFP), one cyan (mCerulean), and one RFP (mScarlet-I). All reporters were constitutively expressed from the chromosome (replacing the non-essential amyA locus) [36] and evaluated based on the excitation and emission optima determined using plasmid versions of the reporter constructs. First, we characterized the brightness of the chromosomally encoded versions via endpoint measurements in relation to the background fluorescence of the WT S. enterica (Fig. 1A). Among the tested FPs, mScarlet-I showed the highest signal over background fluorescence with an 80-fold higher signal relative to wild type levels (Fig. 1B). For both FPs in the green spectrum, mNeonGreen showed lower brightness than eGFP; however, the signal over the background fluorescence was slightly enhanced for mNeonGreen. Of the four tested FPs, mCerulean showed the lowest ratio compared to the WT, with only an eightfold increase in signal over background.
After the general characterization of the FPs, we next focused on evaluating mCerulean, eGFP, mNeonGreen, and mScarelt-I in greater depth. For this purpose, we first manipulated the stability of the reporters to improve their applicability in monitoring rapidly changing cellular processes.
SsrA-tag mediated degradation of fluorescent proteins to enhance reporter dynamicsBacteria encode sophisticated protein control and quality systems to avoid synthesis of error-prone polypeptides. In Escherichia coli (E. coli) and S. enterica, the SsrA-tag system is utilized to release stalled ribosomes of transcripts lacking stop codons [37]. The ssrA RNA molecule, which functions as a transfer and messenger RNA (tmRNA), binds to stalled ribosomes. Upon binding of the tmRNA, a short open reading frame encoding the SsrA tag is translated and added to the nascent peptide. This leads to translation termination by the stop codon within ssrA and release of the stalled ribosome [38]. In turn, the peptide is now primed to be recognized by protease complexes including FtsH, Lon, Tsp, ClpAP, and most prominently ClpXP, resulting in the degradation of truncated and mistranslated proteins [38, 39]. This system can be used to decrease the stability of the engineered proteins [40]. To explore the application of unstable reporter proteins and tune their temporal resolution capacity, plasmid-based constitutively expressed FPs were fused to different SsrA-tags with varying specificity to the degradation complexes (Fig. 2A, B). This approach was previously described with degradation tags containing the amino acid sequences: AANDENYALVA (abbreviated as LVA), AANDENYAAAV (AAV), and AANDENYAASV (ASV) that displayed decreasing degradation efficiency in E. coli [41]. The variation in degradation efficiency depends on the affinity of the tag to host proteases. Subsequently, a high affinity to the protease mediates fast degradation, whereas a lower affinity results in slow degradation dynamics. Importantly, when comparing the degradation of identical SsrA-tagged GFP constructs between E. coli and Pseudomonas putida, the protein stability differed between the two species [41]. Thus, a species-specific characterization of such tags is indispensable. Therefore, we tested the degradation kinetics of representative FPs covering the entire spectrum of visible light using fusions of three variations of the SsrA tag in S. enterica. LVA, AAV, and ASV were individually fused to mCerulean, eGFP, mNeonGreen, and mScarlet-I, and the protein degradation kinetics were determined using a pulse-chase experiment. Translation was inhibited by the addition of chloramphenicol and spectinomycin to bacterial cultures grown to mid-exponential phase (Fig. 2). By comparing the fluorescence intensity of the tagged and non-tagged FPs at the time point of growth arrest (t = 0), we observed that the presence of any SsrA tag increased the steady-state degradation of the FP, resulting in a reduction in the overall reporter signal strength (Fig. 2A). LVA-tagged FPs decreased the signals of mCerulean, eGFP, and mScarlet-I by approximately 50%, whereas the signal of mNeonGreen was reduced by approximately 85% compared to the untagged FPs. The AAV and ASV tags decreased the reporter strength of mCerulean, eGFP, and mScarlet-I by approximately 40% and 10%, respectively. Notably, AAV-tagged mNeonGreen depicted a reduction of about 70% and ASV-tagged mNeonGreen of 40% compared to the untagged FP (Fig. 2A). After the addition of antibiotics and inhibition of de novo protein synthesis, we next investigated the tag-dependent degradation kinetics of the FPs by monitoring changes in the fluorescence intensity over a period of 3 h. Subsequently, we plotted the ratios from the fluorescence signals obtained of biological triplicates (Fig. 2B and C; raw data Figure S4). Also, we determined the time at which 25% (T25) or half of the signal (T50) was reached compared to non-tagged FPs (Table S2). Consistent with steady-state degradation levels, we observed that LVA-tagged proteins showed the strongest and fastest degradation kinetics (Fig. 2B). The AAV and ASV tags resulted in degradation kinetics between non-tagged and LVA-tagged FPs. Strikingly, for mNeonGreen but not for the other tested FPs, we did not observe differences in the degradation kinetics between the LVA and AAV tags (Fig. 2B). For mScarlet-I, we detected an increase in the fluorescence signal for all constructs after inhibition of translation. The most likely explanation for this observation is a delayed fluorophore maturation after protein synthesis. A similar yet less pronounced effect was observed for eGFP (Fig. 2B), and the addition of any SsrA degradation tags reduced the overall signal strength for both FPs, demonstrating their general functionality in protein degradation.
Fig. 2
Evaluation of SsrA degradation tags that modulate the stability of FPs. A Relative fluorescence intensity of tagged FPs compared with their non-tagged counterparts. The tags cause a mild to moderate drop of maximum FP intensity that correlate with their affinity to be degraded. B + C Ratios of pulse-chase experiments (for the original data see Figure S3) to observe degradation speed over a time of 3 h after inhibition of de novo translation. Non-tagged FPs remain stable or show an increase in fluorescence intensity, due to maturation time of the FPs. The tagged FPs decrease in intensity at different speeds reflecting their degradation efficiency. For genome based tagged FPs, only fluorescence was detected for the ASV tag (see main text)
To the best of our knowledge, previous investigations concerning the degradation kinetics of SsrA tags have only been performed using plasmid-based reporters, which motivated us to characterize the degradation kinetics of genome-encoded FPs (Fig. 2A, C). Surprisingly, for genome-encoded FPs, no fluorescence signal was detected for strains expressing FPs fused to tags mediating fast or intermediate degradation (LVA, AAV), independent of protein synthesis arrest, indicating efficient degradation before chromophore maturation (Figure S3). However, we observed a fluorescence signal in strains expressing proteins fused to the ASV tag, which mediated the slowest degradation within our set of degradation tags. When comparing the fluorescence of the non-tagged and tagged reporters at the time of antibiotic addition, mNeonGreen and mScarlet-I exhibited approximately 40% reduction in fluorescence intensity (Fig. 2A). After inhibition of de novo protein translation, the levels of mNeonGreen-ASV decreased constantly, whereas for the non-tagged protein, the signal remained stable until the end of the experiment (Fig. 2C). Similar to plasmid-based experiments, mNeonGreen displayed the fastest degradation with mNeonGreen-ASV, depicting a signal reduction to 50% within one hour after growth arrest. Three hours after translation inhibition, the fluorescent signal of mNeonGreen-ASV was reduced to 20% relative to non-tagged mNeonGreen. In comparison, the mScarlet-I signal initially increased after translation inhibition in the ASV-tagged and non-tagged FPs, as we previously observed for the plasmid-based version and can be explained by a maturation delay of the already fully translated mScarlet-I prior to the addition of antibiotics (Fig. 2C and S3). mScarlet-I displays a significantly longer maturation time than mNeonGreen, eGFP, or mCerulean [26]. Despite this maturation delay, the levels of mScarlet-I-ASV decreased by approximately 30% compared to the non-tagged reporter protein at the end of the experiment. Similar results were observed for the other two reporter proteins, mCerulean-ASV and eGFP-ASV, in which the fluorescence levels constantly decreased after translation inhibition (Fig. 2A and C). After 3 h of incubation, a decrease in fluorescence of approximately 40% and 60% was observed for mCerulean-ASV and eGFP-ASV, respectively. In summary, fusions of SsrA degradation tags to FPs decreased the stability of chromosomally encoded reporters, rendering these improved reporters more sensitive with an increased temporal resolution.
Since the proper function of the degradation tags is dependent on the activity of the host proteases, we next addressed how the deletion of ClpXP, a global protease complex, impacts the degradation kinetics of the reporter FPs [38, 39]. For this, we separately transformed the plasmid-encoded eGFP reporter variants into a S. enterica ∆clpXP strain, performed a pulse-chase degradation experiment, and monitored reporter activity over time after translation arrest (Fig. 3A). As expected, the absence of ClpXP resulted in no degradation of ASV- and AAV-tagged eGFP compared to the non-tagged eGFP. Interestingly, we detected some degradation of the LVA-tagged construct (Fig. 3A). These findings suggest that ASV- and AAV-tagged proteins are only recognized and degraded by ClpXP, whereas other proteases such as FtsH or Lon are capable of degrading LVA-tagged proteins in the absence of ClpXP [42, 43].
Fig. 3
Effect of ClpXP on the SsrA-mediated degradation dynamics of tagged FPs. A Pulse-chase experiment for three hours of plasmid-based eGFP with degradation tags or non-tagged in a ∆clpXP S. enterica background. B Pulse-chase experiment of genome-based mNeonGreen expression either performed in exponential phase (3 h) or stationary phase (5 h)
In addition to the general importance of ClpXP for degradation of FP reporters, we next investigated how varying protease levels affect the activity of a SsrA tagged reporter construct at different growth phases. Based on the SalCom RNA sequencing database, we determined that clpXP transcription levels peak in mid-exponential phase and decrease substantially in early- and late-stationary phase [44]. Accordingly, we determined the degradation kinetics of a genome-based mNeonGreen reporter with and without ASV tag after 3 h (exponential phase) and 5 h (stationary phase) of growth (Fig. 3B). Interestingly, we did not observe any differences in the degradation kinetics, suggesting that at both tested timepoints sufficient levels of ClpXP were available to degrade the ASV tagged reporter (Fig. 3B).
Tagged fluorescent proteins enhance temporal resolution of transcriptional fusionsIn order to investigate the applicability of SsrA-tagged FP reporters for monitoring gene expression dynamics, we next probed the activity of the well-studied constitutive promoter PrpsM that natively drives the expression of the ribosomal protein S13 throughout various growth phases [45, 46]. Based on the above-mentioned global RNA sequencing database, we predicted strong and steady FP reporter levels expressed from PrpsM ranging from early exponential phase into early stationary phase [44]. Subsequently, in late stationary phase, we predicted a notable drop of promoter activity due to the stringent response [47]. Accordingly, we generated a transcriptional fusion of PrpsM to mNeonGreen with or without ASV tag in the amyA locus of S. enterica and followed growth as well as reporter signal over a time course of eight hours (Fig. 4). Until late exponential phase (about 3 h after start of the experiment), we did not observe substantial differences in reporter activity between the tagged and untagged mNeonGreen variant. However, as soon as the bacteria entered stationary phase, the downregulaton of PrpsM was only observed for the ASV-tagged mNeonGreen variant (Fig. 4). The non-tagged mNeonGreen construct displayed a constant signal over the entire time of the experiment, thereby masking the growth phase dependent transcriptional downregulation of PrpsM (Fig. 4). This observation underscores the necessity of incorporating degradation tags to fully reveal promoter dynamics in vivo.
Fig. 4
Transcriptional profile of tagged and non-tagged FP reporters. Growth and fluorescence intensity of strains harboring the ribosomal S13 promoter (PrpsM) fused to mNeonGreen or mNeonGreen-ASV were monitored in a plate reader over time. Growth and fluorescence measurements were performed every 30 min for eight hours. Means and standard deviation derive from at least three independent replicates and are represented by error bars for growth or shadowed space for fluorescence
Next, we probed if the addition of degradation tags affects investigations of population heterogeneity. For this we analyzed the heterogeneity of genome-based and plasmid-based transcriptional fusions of PrpsM to mNeonGreen with or without tags on the single cell level. As proxy for heterogeneity in gene expression, we determined the coefficient of variation (CoV) in fluorescence intensity of cultures grown to exponential phase using fluorescent microscopy (Fig. 5). Surprisingly, a plasmid-based mNeonGreen lacking a degradation tag depicted a CoV of 0.31, which doubled for the LVA tagged version. Lower CoV values indicate less variation of measured fluorescence within a population set relative to the mean [48]. Satisfactorily, both the plasmid-based constructs harboring the ASV or AAV tag decreased the heterogeneous expression of mNeonGreen driven by PrpsM compared to the non-tagged counterpart. In our approach, this could indicate that rapid degradation of a reporter fusion impacts cell-to-cell heterogeneity, and moderately strong degradation tags result in a more homogenous signal. However, to draw a general conclusion in other contexts, this would need further verification.
Fig. 5
Determination of cell-to-cell variation using degradation tagged and non-tagged mNeonGreen transcriptional reporters. A Coefficient of variation histograms of mNeonGreen driven by PrpsM both on plasmid- as well as genome-based fused to one of the SsrA degradation tags or non-tagged. B Exemplary fluorescence microcopy pictures of the constructs in A. Scale bar is 2 µm and the fluorescence signal intensities were adjusted separately for plasmid and genome-based images, respectively
Interestingly, comparing the CoV of genome-based mNeonGreen expression depicted only a neglectable difference between tagged and non-tagged reporters. Satisfactorily, this reflects well the trend observed for also the plasmid-based reporters.
Although transcriptional reporters remain a major part of the application portfolio of FPs, translational FP fusions are also frequently used to investigate protein localization and dynamics. Therefore, we next set out to demonstrate the applicability of FPs as translational fusions in life cell studies, as well as fixed samples, using fluorescence and super-resolution microscopy.
Fluorescence proteins to study protein localisation and dynamicsAs a proof of concept, we fused representative FPs to a C-ring protein of the flagellar basal body in S. enterica. For this, we chose FliG, a protein part of the flagella type-III secretion system (fT3SS), which interacts with the stator units MotAB to drive rotation of the flagellum [49,50,51]. N-terminal, translational fusions were constructed at the native fliG locus (Fig. 6A). To quantitatively evaluate the performance of FPs as proxies for FliG localization, we systematically analyzed the signal to background in cells expressing each FP fusion using epifluorescence microscopy (Fig. 6B). The signal to background ratio indicated that while there was variability among the FPs, several candidates produced signals significantly above the background levels, which is suitable for localization imaging studies. For instance, sfGFP, mNeonGreen, and mScarlet-I exhibited higher signal to background values compared to mRuby3 or mNeptune2 fusions. However, fluorescence microscopy imaging revealed distinct localization patterns of the FPs corresponding to FliG complexes assembled in C-rings, and we observed that visualization of blue excitable FPs (sfmTurquoise2ox, mNeonGreen, sfGFP) was more easily detectable than orange/red excitable FPs and would therefore be more suitable for foci fluorescence observations (Fig. 6C). Successful fusion and functional expression with the native POI locus require assessment of the functionality of the newly engineered protein. In the case of FliG, we assessed the impact of the fusions by confirming the presence of flagellar filaments and measuring overall motility. Although all fusions were flagellated, motility was affected differently depending on the fusion (Figure S4), indicating that the fusions did not prevent the secretion of flagellar subunits, but did affect interaction with stator units and therefore flagellar rotation [50]. We next utilized Stimulated Emission Depletion (STED) super-resolution microscopy to visualize mNeonGreen-FliG with a higher resolution (Fig. 6D). We observed substantially more distinct FliG foci and reduced background using STED compared to confocal laser scanning microscopy (CLSM). Although FPs in general display lower photostability and are more prone to photobleaching than organic dyes, such as the HaloTag TMR ligand [52], the increased resolution observed here demonstrates that if using a FP is inevitable, mNeonGreen can be applied for STED microscopy in bacteria. Importantly to note is that recent engineering has brought forward several novel green emitting FPs, such as StayGold and derivates of the same, which could be explored for super-resolution microscopy approaches [53,54,55].
Fig. 6
Visualizing cellular structures in bacteria using translational FP fusions. A Schematic representation of the bacterial flagellar motor highlighting the position of the FliG protein within the basal body structure. Figure adapted from Halte et al., 2021. [56]. B Histogram showing the signal-to-noise ratio of the FliG fluorescence foci of different fluorescent protein fusions (live cell), indicating the efficiency of each fluorophore for fluorescence focus observation. C Representative epifluorescence microscopy images displaying the distribution of the FliG protein fused to various fluorescent proteins within the bacterial cells (scale bar = 2 µm). D Representative confocal laser scanning microscopy (CLSM) images and super-resolution STED (Stimulated Emission Depletion) microscopy image of FliG-mNeonGreen fusion, showcasing the enhanced resolution of FliG protein localization using STED (scale bar = 1 µm)
Collectively, these results underscore the versatile applicability of FPs also for the study of protein localization and functions in Gram-negative bacteria.
留言 (0)